
جدول محتوا
مقدمه
فرآیند های اپی ژنومیک با تغییر ساختار فضایی کروماتین به کنترل بیان ژن می پردازند. به طوری که ژن هایی که بیان فعال داشته باشند عموما در نواحی در دسترس کروماتین قرار گرفته اند درحالی که ژن هایی که از نظر بیان خاموش باشند اغلب در مناطق غیر قابل دسترس کروماتین جای دارند. بر همین اساس به مجموعه تغییراتی که در بخش DNA یا پروتئینی کروماتین رخ می دهد و ساختار آن را تحت تاثیر قرار می دهد، اپی ژنوم گفته می شود. در همین خصوص، استیلاسیون توالی اسیدهای آمینه هیستون ها همبستگی مثبتی با افزایش بیان ژن داشته و بالعکس، نواحی متیله شده هیستون عموما به صورت نقاط فشرده کروماتین در نظر گرفته شده و از نظر بیان ژن ها غیر فعال می باشد.
به طور مشابه، متیلاسیون ریشه های سیتوزین در توالی DNA کروماتین نیز عموما سطح دومی از سرکوب بیان ژن را به دنبال دارد و الگوی بیان ژن را تعیین می کند. براساس آنچه پیشتر گفته شد، تکنیک های بررسی اپی ژنوم معمول با هدف بررسی متیلاسیون- استیلاسیون هیستون ها و نیز متیلاسیون DNA انجام می پذیرند. در این مقاله به صورت اجمالی با برخی فناوری های مرتبط با بررسی متیلاسیون DNA آشنا خواهیم شد. تکنیک های بررسی متیلاسیون DNA بر دو رویکرد کلی مبتنی هستند که شامل conversion Bisulfite و Immunoprecipitation DNA Methylated می شود. در روش conversion Bisulfite چندین پلتفرم متفاوت توانایی بررسی متیلاسیون DNA تیمار شده با بی سولفیت سدیم را فراهم می کنند. طی این فرآیند، سیتوزین های غیر متیله دچار دآمیناسیون شده و به یوراسیل تبدیل می شوند. تکنیک های پایین دست با شناسایی یوراسیل در هر نقطه از DNA تیمار شده و مقایسه با نمونه تیمار نشده پی به متیله یا غیر متیله بودن نوکلئوتید سیتوزین می برند. علاوه بر این، تکنولوژی Immunoprecipitation DNA Methylated محققین را قادر می سازد تا با بررسی گسترده تغییرات ژنومی به الگوهای متیلاسیون DNA پی ببرند. برخلاف تیمار بی سولفیتی، در این
روش از آنتی بادی های اختصاصی علیه اهداف DNA متیله استفاده می شود تا این نواحی جدا شده و برای بررسی بیشتر از طریق تکنولوژی هایی با قدرت تفکیک بالاتر همانند microarray و تکنیک های توالی یابی مورد آنالیز قرار بگیرد.
از سوی دیگر، تکنیک های بررسی برهم کنش های DNA -پروتئین مسیرهای شناسایی اصلاحات هیستونی را روشن می سازد. برهم کنش بین DNA و پروتئین ها) به ویژه هیستون ها و فاکتورهای رونویسی (اغلب به کمک تکنیکی تحت عنوان immunoprecipitation chromatin صورت می گیرد. در این روش با کمک پنلی از آنتی بادی های مختلف مجموعه های DNA و پروتئین های متصل به DNA شناسایی و اهمیت این پروتئین ها در ساختار کروماتین و بیان ژن مورد بررسی قرار می گیرد.
پروژه های مرتبط با اپی ژنوم
پروژه های بین المللی بررسی اپی ژنوم انسانی عموما با تمرکز بر متیلاسیون توالی DNA به دنبال راهی برای الگوسازی از اپی ژنوم بیماران و مخصوصا افراد سرطانی می باشند. در ادامه لیستی از پروژه های مرتبط با اپی ژنوم گنجانده شده است:
• Human Epigenome Project (HEP): http://www.epigenome.org
• Cancer Genome Project (CGP): http://www.sanger.ac.uk/genetics/CGP
• The Cancer Genome Atlas (TCGA): http://cancergenome.nih.gov
• NIH Roadmap Epigenomics Program: http://www.roadmapepigenomics.org
• International Cancer Genome Consortium (ICGC): http://www.icgc.org
• International Human Epigenome Consortium (IHEC): http://www.epigenomenoe.net/ihec
• Epigenome Network of Excellence: http://www.epigenome-noe.net
• Treat 1000 Project: http://www.treat1000.org
واژه اپی ژنتیک اولین بار توسط Waddington Conrad به کار گرفته شد و تا به امروز در تعریف دستخوش تغییراتی شده است. اپی ژنتیک هم اکنون به همه تغییرات غیر قابل توارث محتوای ژنوم گفته می شود که بدون تغییر در توالی بازهای آلی موجود در DNA سبب تغییر میزان بیان ژن می گردد. اهمیت این فرآیند زمانی مشخص می شود که بدانیم تمامی فرآیند های زیستی همچون تمایز سلولی، تکامل و اکولوژی موجود تحت تاثیر بیان ژن می باشد. بنابراین، اختصاصی بودن اپی ژنوم هر فرد پلی منحصر به فرد بین ژنوتیپ (توالی ژنتیکی) و فنوتیپ (شکل و عملکرد) سلول ها و ارگان های مختلف برقرار می کند. به بیان دیگر، تعامل ژنتیک و اپی ژنتیک پیغام تنظیمی اصلی برای سرنوشت یک سلول و در ابعاد بزرگتر برای یک جاندار می باشد. بر همین اساس، پایداری ژنتیک فرد به همراه انعطافی که در اپی ژنتیک وجود دارد، الگوهای متعدد و بسیار پیچیده ای از تنظیم بیان ژن در پاسخ به محرک های داخلی و خارجی فراهم می آورد. از آنجاییکه گروه متیل و استیل در کنترل بیان ژن های مختلف دارای نقش اساسی می باشند، متیلاسیون و استیلاسیون باز های آلی DNA دو مکانیسم اصلی تغییرات اپی ژنتیک در پستانداران می باشند که به طور گسترده در حوزه بیان ژن مطالعه شده اند.
در سلول ها، متیلاسیون به واسطه فعالیت سه گروه از آنزیم ها که فرآیند متیلاسیون، دمتیلاسیون و خوانش متیلاسیون را برعهده دارند صورت می پذیرد. از میان آنزیم هایی که فرایند متیلاسیون را کنترل می کنند می توان به انواع DNA متیل ترانسفرازها نظیر DNMT1 و DNMT 3b/3a) اشاره داشت که گروه متیل را از منبع آمینو اسید متیونین به نوکلئوتید سیتوزین غیر متیله بویژه در نواحی غنی از سیتوزین-گوانین (تحت عنوان جزایر CpG) منتقل می کنند. در عوض، خانواده آنزیمی family،of ten-eleven translocation (TET) methylcytosine dioxygenase (نظیر TET3 و TET-2 ،TET-1) گروه متیل را از نوکلئوتید سیتوزین متیله جدا کرده و منجر به حذف آن می شوند. براساس شواهد موجود، تمامی آنزیم های فوق جهت صحت فرآیندهای زیستی تکامل و تمایز سلولی ضروری می باشند و فعالیت نامعمول آنها در بسیاری بیماری ها گزارش شده است. افزایش متیلاسیون DNA عموما با اثرات مهاری همانند فراخواندن پروتئین های متصل شونده به متیل، مهار انواعی از فاکتور های رونویسی و تغییرات کروماتین همراه می باشد. امروزه دقت، حساسیت، سادگی و هزینه روش های بررسی متیلاسیون DNA بسیار متنوع است. بنابراین انتخاب استراتژی درست بررسی متیلاسیون ضروری می باشد.
روش های مختلف بررسی متیلاسیون DNA
ارزیابی الگوی متیلاسیون DNA در سطح نوکلئوتیدها از جمله روش های بررسی پروفایل اپی ژنتیک می باشد . تا جایی که بسیاری از بیماری های پیچیده همچون سرطان، بیماری های خود ایمن و نورودژنراتیو، همراه با تغییرات گسترده الگوی متیلاسیون DNA همراه هستند. به دلیل آنکه جهت سنجش متیلاسیون DNA از روش های رایج نظیر PCR معمولی نمی توان استفاده کرد، محققان به منظور قابل شناسایی کردن نوکلئوتیدهای متیله با استفاده از PCR ،از ترکیبات شیمیایی مختلفی استفاده کرده اند. به طوریکه، ترکیبات مورد استفاده موجب تغییر توالی نوکلئوتیدی فقط در نواحی متیله می شود. یکی از قدیمی ترین روش ها که امروزه نیز به طور گسترده در آزمایشگاه استفاده می شود، روش تیمار DNA با بی سولفیت می باشد. تیمار DNA با بی سولفیت سبب می شود که بازهای غیر متیله سیتوزین گروه آمین خود را از دست بدهند و به باز یوراسیل تبدیل شوند و پس از تکثیر به روش PCR یوراسیل ها به تیمین تبدیل می شوند. درصورتی که باز سیتوزین به صورت متیله باشد تیمار بی سولفیت اثری بر روی این باز نخواهد داشت و پس از PCR نیز به صورت سیتوزین باقی می ماند. از آنجا که همه تکنیک های بررسی متیالسیون نواحی CpG نیازمند تیمار DNA با بی سولفیت می باشند، براساس گستردگی نواحی DNA مورد بررسی، تکنولوژی های مختلف گاهی ناحیه کوچکی از ژنوم را بررسی می کنند یا شاید حتی بر کل ژنوم تمرکز داشته باشند . از همین رو روش های موجود به سه دسته با توان عملیاتی پایین ، متوسط و بالا تقسیم بندی می شوند. در ادامه تمرکز مقاله حاضر عمدتا بر تکنیک های مبتنی بر آنزیم های محدودالاثر، تیمار بی سولفیتی و نهایتا فناوری های مبتنی بر میل پیوندی خواهد بود.
1 .تکنیک های مبتنی بر آنزیم های محدودالاثر
آنزیم های محدود الاثر به گروهی از آنزیم های باکتریایی اطلاق می شود که توالی نوکلئوتیدی خاصی را شناسایی کرده و سپس باند دی سولفید را هیدرولیز می کنند. برخی از این آنزیم ها همانند HpaII و SmaI به متیلاسیون توالی های DNA به ویژه در نواحی غنی از CpG حساس می باشند. به عنوان مثال، در مورد HpaII توانایی برش سیتوزین متیله در توالی CCGG موجود در جزایر CpG گزارش شده است. برخی از آنزیم های مختلف محدودالاثر توالی یکسانی را شناسایی می کنند و از این رو ایزوشیزومر خوانده می شوند. آنزیم ایزوشیزومریک MspI علی رغم شناسایی توالی CCGG، قادر به برش توالی متیله نمی باشد. بنابراین با استفاده از ترکیبی از ایزوشیزومر ها، به راحتی میتوان الگوی متیلاسیون را در نواحی خاصی از DNA مورد بررسی قرار داد. در ابعاد بزرگتر یا اصطلاحا در مطالعات توان بالا عموما از آنزیم محدودالاثر دیگری بنام McrBC استفاده می شود که با حساسیت پایین تر نسبت به آنزیم های یاد شده تقریبا قادر به شناسایی همه نواحی متیله جزایر CpG می باشد. در نتیجه، انتخاب نوع آنزیم براساس اختصاصیت و حساسیت سبب شده روش های متنوعی از نظر گستره بررسی متیلاسیون DNA با استفاده از آنزیم های محدودالاثر ایجاد و توسعه یابد.
2 .تکنیک های مبتنی بر میل پیوندی پروتئین متصل شونده به متیل و آنتی بادی
مطالعات مختلفی حاکی از توانایی اتصال برخی پروتئین ها، به ویژه فاکتور های رونویسی، به گروه های متیل موجود در رشته DNA می باشند. چنین ویژگی در برخی از آنتی بادی ها نیز گزارش شده و به عنوان تیماری جهت افتراق سیتوزین های متیله ژنوم به کار گرفته شده اند. متدهای مبتنی بر این فناوری در ابعاد گسترده و کوچک مطالعات ژنومی استفاده شده اند و به دلیل کارایی بالا و سرعت انجام فرآیند، محققان را مجاب کرده است که جهت تشخیص متیالسیون در ادامه ی array-hybridization PCR مورد و حتی توالی یابی نسل جدید (NGS) استفاده قرار گیرند. از بارزترین معایب روش های مبتنی بر اتصال پروتئین به سیتوزین متیله آن است که پروتئین های فوق قادر به تشخیص افتراقی وضعیت متیلاسیون در جزایر CpG یا در یک نوکلئوتید CpG نمی باشند.
3 .تکنیک های مبتنی بر تیمار DNA با بی سولفیت
برای اولین بار Frommer و همکارانش در سال ۱۹۹۲ موفق شدند طی تیمار DNA با بی سولفیت سدیم سبب دآمیناسیون و سپس متیلاسیون سیتوزین های موجود در جزایر CpG شوند. بدین ترتیب سیتوزین دآمینه شده به یوراسیل تبدیل شده و طی همانندسازی در آزمایشگاه به تیمین تغییر می یابد. در مقیاس مولکولی، افزودن سولفونات به سیتوزین موجود در نواحی CpG و غیر CpG سبب القای دآمیناسیون کربن چهارم سیتوزین شده و یوراسیل سولفونات شکل می گیرد. این درحالی ست که سیتوزین هایی که از قبل متیله بودند بدون تغییر باقی می مانند. توجه داشته باشید وقایع ذکر شده تنها بر DNA تک رشته رخ می دهد و بنابراین دناتوره کردن DNA حین تیمار با بی سولفیت سدیم ضروری می باشد. همچنین، کیفیت DNA استخراج شده، مقدار و pH واکنش یاد شده از جمله مهم ترین فاکتور های دخیل در فرآیند شیمیایی فوق می باشد و دآمیناسیون ناقص DNA از اساسی ترین معضلات این روش می باشد. این روش ساده اساس بسیاری از متدهای بررسی متیلاسیون مثل MSP ،PCR time-real pyrosequencing ،HRM ،BSP می باشد که در ادامه با توضیح بیشتری شرح داده می شوند، همچنین، این روش همراه با تکنولوژی میکرواری و توالی یابی نسل جدید جهت بررسی متیلاسیون کل محتوای ژنتیکی موجود استفاده می شود.
روش های مبتنی بر میکرواری
در سال ۱۹۹۵، واژه میکرواری در یکی از مقالات علمی به چاپ رسید و به بررسی بیان چندین ژن به صورت همزمان اشاره داشت. فناوری اری و میکرواری به عنوان یک پیشرفت در روش های بررسی بیان ژن معرفی شده است. بر همین اساس، دانشگاه استنفورد در سال ۱۹۹۷ اولین چیپ میکرواری جهت توالی یابی کل اگزوم را تهیه و روش تولید را برای همگان منتشر کرد. تا سال ها این روش تجاری سازی نشده بود تا آن که با پیشرفت پرینترهای روباتیک، امکان تولید گستره و درنتیجه توسعه چیپ های میکرواری فراهم شد. این فناوری از آنجا حائز اهمیت می باشد که هر چه تعداد بیشتری از قطعات الیگونوکلئوتیدی پروب به سطح چیپ های شیشه ای اتصال یابد، امکان آنالیز گسترده تر محتوای ژنومی فراهم می شود.
در ادامه اهداف گسترده تری برای تکنولوژی میکرواری مهیا شد و اولین تلاش های استفاده از اری برای بررسی متیلاسیون و به طور کلی بررسی اپی ژنتیک قطعات DNA در سال ۲۰۰۲ صورت گرفت. این روش تحت عنوان Mehtylation specific oligonucleotide microarray (MSO microarray) توسط Tim Hui-Ming به جامعه آزمایشگاهی معرفی شد.
فناوری میکرواری از جمله فناوری های در حال توسعه بررسی بیان ژن های مختلف به طور همزمان می باشد. این روش به طور خلاصه شامل قرار دادن توالی هزاران قطعه ژن مختلف بر روی اسلاید های شیشه ای تحت عنوان چیپ ژنی می باشد. در نتیجه با مجاور کردن نمونه DNA یا RNA مجهول با چیپ یاد شده، قطعات مکمل نمونه مجهول به قطعات ژنی روی اسلاید اتصال یافته و نور از خود ساطع می کنند. شناسایی نور از هر قطعه چیپ توسط آشکارسازها بدان معناست که ژن مورد نظر در نمونه اضافه شده به چیپ بیان داشته است.
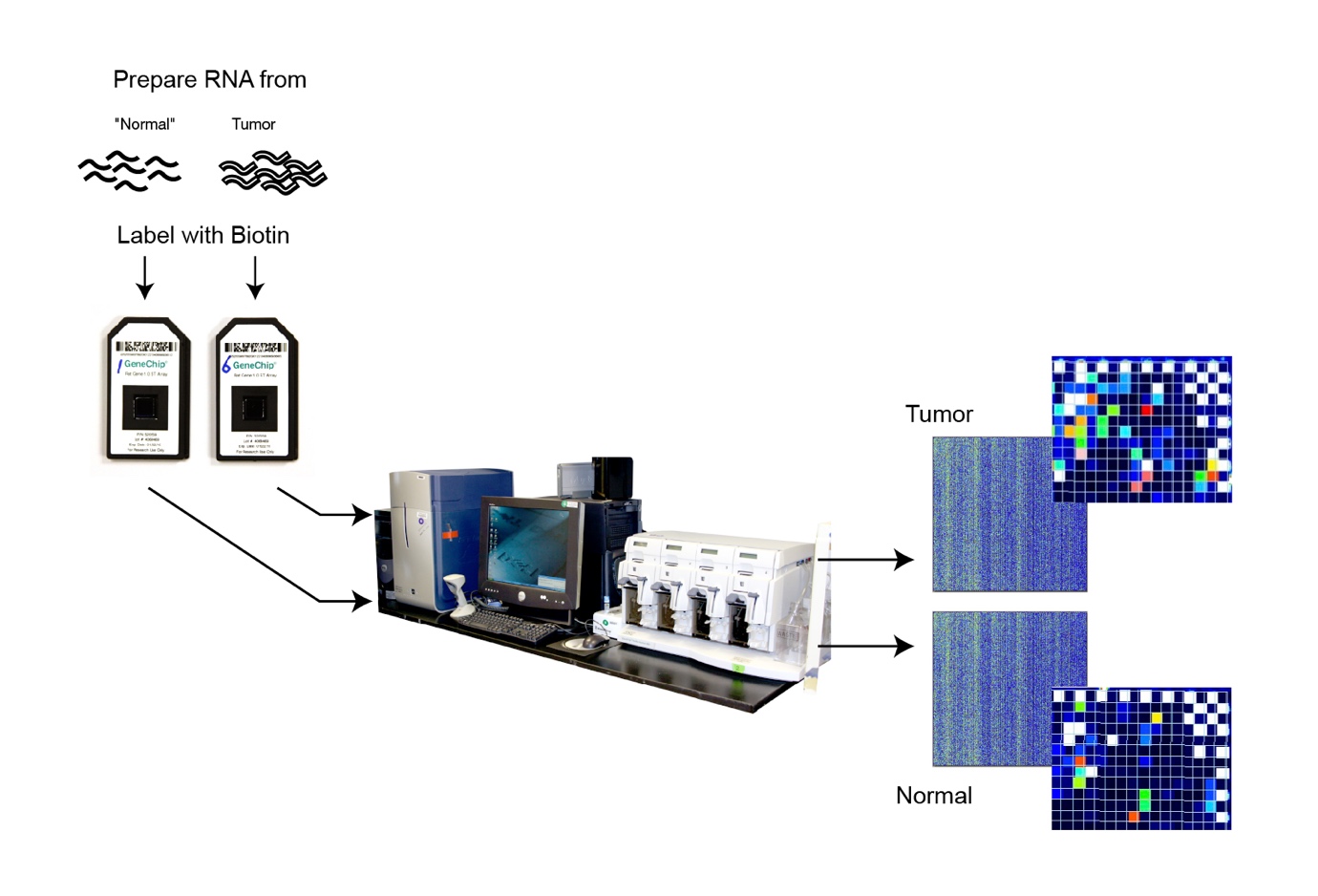
ساختار چیپ
فناوری میکرواری و چیپ های تجاری موجود برای آن براساس هدف آزمایشگاهی مورد نظر بهینه سازی شده اند. مثلا تکنولوژی Bead Array شرکت Illumina جهت بررسی متیلاسیون قطعات DNA تیمار شده با بی سولفیت طراحی و تولید شده است. در ادامه اساس کار این فناوری توضیح داده شده و ساختار چیپ مورد بررسی قرار خواهد گرفت.
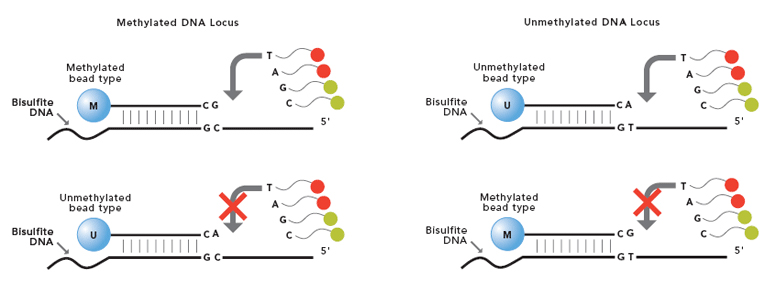
تکنولوژی میکرواری ایلومینا، تحت عنوان Infinium methylation assay، از میکروبید های سیلیکا استفاده می کند که در سطح خود حاوی صدها هزار تا میلیون ها توالی ژنوتیپی است که برای یک فرد قابل بارگذاری شدن می باشد. در روند تولید این چیپ ها، بید ها ابتدا با دقت بسیار بالایی در چاهک های کوچکی قرار داده شده و سپس چندین نسخه از پروب الیگونوکلئوتیدی که ناحیه خاصی از ژنوم را هدف قرار می دهد، روی بید بارگذاری می شود. همزمان با عبور قطعات DNA نشاندار شده با رنگ های فلوئورسنت از روی بیدها، هر پروب به توالی مکمل خود در نمونه DNA اتصال می یابد. حال به کار بردن لیزر جهت برانگیختن رنگ فلوئورسنت، در صورت وجود هیبرید پروب – DNA نمونه مجهول، سیگنال رنگی آزاد شده که براساس شدت سیگنال، میزان بیان یا متیله شدن سیتوزین موجود در DNA مشخص می گردد. قابل توجه آنکه، در فرآیند آزمایش فوق (بررسی متیلاسیون) دو نوع بید طراحی می شود. به طوری که یک بید در توالی پروب خود واجد نوکلئوتید آدنین بوده و به باز تیمین (یا به عبارتی یوراسیل ایجاد شده بعد از متیلاسیون سیتوزین اتصال می باید) و توالی پروب موجود در سطح بید دیگر واجد گوانین بوده و به سیتوزین غیر متیله اتصال می یابد.
پروتکل آزمایشگاهی
1. تکثیر قطعه DNA
اولین گام در پروتکل آزمایشگاهی بررسی متیلاسیون براساس فناوری microarray شرکت ایلومینا تکثیر قطعه DNA نمونه مجهول می باشد. برای اینکار ابتدا کافی ست محلول مورد نیاز تکثیر را از فریزر خارج کرده و بعد از ذوب شدن به نمونه های DNA تخلیص شده اضافه کرد. هدف از اینکار تکثیر کامل ژنوم موجود در نمونه مورد بررسی می باشد تا برخلاف PCR، محدود به مناطقG-C نباشد. بدین ترتیب نمونه اولیه ۱۰۰۰ برابر تکثیر شده و میزان کافی از DNA برای میکرواری مهیا می شود.
2. قطعه قطعه شدن DNA
همانند مرحله قبل با خارج کردن محلول fragmentation و ذوب کردن آن در دمای محیط، میزان مورد نیاز از محلول به چاهک های DNA تکثیر شده اضافه می شود تا نمونه ژنومی به اندازه های بهینه جهت هیبریداسیون با پروب قطعه قطعه گردد. آنزیم های اندونوکلئاز موجود در این محلول، DNA را به قطعات ۳۰۰ الی ۶۰۰ جفت باز خرد می کنند.
3. رسوب دهی
در این مرحله، ابتدا محلول رسوب دهنده از یخچال خارج و به دمای محیط رسانده می شود. همچنین، محلول مورد نیاز برای حل کردن نمونه از فریزر خارج و حداقل یک ساعت جهت ذوب شدن و به دما رسیدن در محیط قرار می گیرد.
پیش از هیبریداسیون نمونه DNA، تخلیص ژنوم به کمک ایزوپروپانول موجود در محلول precipitation صورت می گیرد. در نتیجه این محلول نیز به چاهک های محتوی نمونه اضافه می گردد. سپس با یک مرحله سانتریفوژ کردن در دمای ۴ درجه سانتی گراد، رسوب DNA به دست آمده و سوپ رویی به روش Decant کردن دور ریخته می شود.
4. حل کردن نمونه
پس از آنکه رسوب تخلیص شده DNA به دست آمد، زمان حل کردن نمونه در محلول هیبریداسیون می باشد. این محلول شرایط بهینه مورد نیاز برای هیبریداسیون در سطح بیدها را فراهم می کند.
5. هیبریداسیون نمونه DNA
در این مرحله ابتدا استریپ های محتوی DNA روی یک درپوش با دمای ۹۵ درجه قرار می گیرند تا دو رشته قطعات DNA از یکدیگر باز شوند. سپس پلیت در دمای محیط به مدت ۳۰ دقیقه انکوبه و تحت چرخش قرار می گیرد. در ادامه نمونه DNA از داخل چاهک های استریپ خارج و به سطح اسلاید میکرواری اضافه می شود. اختصاصیت آزمایش در این مرحله و براساس طول پروب، بافر و دمای هیبریداسیون مشخص می گردد. پس از بارگذاری نمونه ها به هر چیپ، اسلاید در محفظه خاص خود قرار گرفته و مجموعه به یک آون با دمای ۴۸ درجه برای مدت 18-16 ساعت انتقال می یابد. در همین حین، محلول های شستشو و coating براساس پروتکل کیت رقیق شده و در دمای منفی ۲۰ نگه داری می شوند. بعد از سپری شدن زمان انکوباسیون، اسلاید از محفظه خود خارج و در دمای اتاق قرار می گیرند.
6. شستشوی بیدچیپ ها
بیدچیپ های خارج شده از محفظه، فورا وارد محلول شستشو شده و در یک قاب پلاستیکی فیکس می شوند.
7. X Stain
ابتدا کافیست محلول های رنگ آمیزی از فریزر خارج شده و در محیط قرار گیرند تا ذوب شوند. وجه تسمیه این مرحله به دو واژه extension و staining باز می گردد. منظور از extension افزودن یک نوکلئوتید نشاندار به هر یک از پروب های موجود در سطح بیدها می باشد تا ژنوتیپ پروب موجود در هر بید (از نظر هموزیگوت و هتروزیگوت بودن براساس قطعه DNA مکمل شده و متصل به آن) از یکدیگر قابل افتراق باشند. این نوکلئوتید برای شناسایی تیمین در نمونه DNA، از نوع دی دئوکسی آدنوزین تری فسفات (ddATP) نشاندار شده با دی نیترو فنل و برای شناسایی سیتوزین غیر متیله از نوع دی دئوکسی گوانوزین تری فسفات (ddGTP) نشاندار شده با بیوتین می باشد. در نتیجه، قطعات DNA موجود در نمونه مجهول اگر از هنگام اتصال به قطعه پروب، هموزیگوت باشند فقط یک نوع سیگنال از خود ساطع خواهند کرد ولی از نمونه های هتروزیگوت طبیعتا دو نوع سیگنال شناسایی خواهد شد.
سپس با یک مرحله شستشو، قطعات DNA که به پروب اتصال پیدا نکرده اند از سطح چیپ شسته می شوند.
هدف دوم از این مرحله Staining می باشد. بدان معنی که سیگنال فلوئورسنت اختصاصی از هر پروب به سمت detector ها روانه شود. برای اینکار رنگ آمیزی ابتدا با streptavidin متصل به فلوئوروکروم سبز آغاز می شود و با افزودن آنتی بادی ضد دی نیتروفنل متصل به فلوئوروکروم قرمز ادامه می یابد.
8. Imaging
به عنوان آخرین گام، چیپ ها در محفظه اسکنر قرار گرفته و پس از دریافت تشعشع لیزر، در هر نقطه شروع به ساطع کردن نور فلوئورسنت سبز یا قرمز و یا ترکیبی از این دو می کنند. Detectorها با شناسایی نور، اطلاعات را به کامپیوتر انتقال و متیله یا غیر متیله بودن قطعه DNA متصل به هر پروب سنجیده می شود.
آنالیز داده
Pipeline در تحلیل داده های ژنومیک به مجموعه مراحل پردازش اطلاعات اشاره دارد که به صورت پی در پی انجام می گیرد و نتایج خروجی یک مرحله به عنوان داده ورودی مرحله بعدی تلقی می شود.
پایپلاین آنالیز داده های خام حاصل از میکرواری بصورت زیر می باشد:
1. کنترل کیفی نمونه
در این مرحله کیفیت نمونه ورودی به آزمایش از طریق کنترل های واجد DNA و کنترل فاقد نمونه DNA سنجیده می شود. بر همین اساس، نموداری از وضعیت بی سولفیته شدن نمونه های DNA و کنترل های آزمایش با کمک نرم افزار GenomeStudio ترسیم شده (پکیج HumMethQCReport) در نرم افزار R نیز برای این کار قابل استفاده می باشد و با یکدیگر مقایسه می شوند.
2. کنترل کیفی پروب
مشابه مرحله قبل عملکرد پروب ها از نظر توانایی اتصال به قطعات DNA هدف سنجیده می شود. به ویژه این موضوع زمانی حائز اهمیت می گردد که نمونه های DNA بی سولفیت که به عنوان کنترل در مرحله قبل استفاده شده اند، وضعیت مطلوبی نداشته باشند و احتمال نقص در پروب ها به دلایل مختلف قوی باشد. یکی از مهم ترین مشکلاتی که در این مرحله ممکن است محققان با آن مواجه باشند این است که پروب به بخش هایی از جزایر CpG اتصال یافته باشد که محل وقوع پلی مورفیسم های تک نوکلئوتیدی (به ویژه SNP) است. به عبارتی درصدی از میزان متیلاسیون یک بخش از DNA ژنومی ممکن است تحت تاثیر ژنوتیپ فرد باشد و محقق تصمیم به حذف لوکوس های مرتبط با پلی مورفیسم داشته باشد. علی رغم آنکه SNP ها به ندرت میزان متیلاسیون را به شدت تغییر می دهند، جهش های سوماتیک که به طور مثال در سلول های سرطانی رخ می دهد، تاثیر چشمگیری در میزان متیلاسیون DNA مورد بررسی دارند.
3. تصحیح پس زمینه
این فرآیند شامل حذف سیگنال های غیر اختصاصی ساطع شده از پلتفرم چیپ می باشد تا از دخالت آن در نتایج حاصله جلوگیری شود.
4. نرمال سازی
در این مرحله نیز منابع تجربی نویز های تصادفی که به دلایل تکنیکال یا سیستمیک در فناوری میکرواری هر چیپ وجود داشته باشد حذف می شوند تا از ایجاد نتایج منفی کاذب جلوگیری شود. بنابراین، برای نرمال سازی دو رویکرد اتخاذ می گردد:
i. Between-array normalization: جهت حذف نویز های تکنیکال بین نمونه های بارگذاری شده روی اری های مختلف
ii. Within-array normalization: جهت تصحیح شدت بایاس ایجاد شده در هر گروه مورد مطالعه به وسیله رنگ های مورد استفاده
توجه داشته باشید که بسیاری از روش های نرمالیزه کردن که برای میکرواری با هدف بررسی بیان ژن به کار می رود، در میکرواری با هدف بررسی میزان متیلاسیون کاربردی ندارد. از طرفی متیلاسیون به طور یکسان در همه بخش های DNA رخ نمی دهد و جزایر CpG از این نظر غنی تر هستند. بنابراین همواره نمودار های توزیع متیلاسیون از یک چولگی برخوردارند. درنتیجه در مرحله نرمالیزاسیون تا حد امکان سعی می شود انحراف طبیعی که در توزیع متیلاسیون وجود دارد، عاری از نویز ناخواسته باشد.
5. همگام سازی پروب نوع یک و دو
از موضوعات مهم دیگر در چیپ های میکرواری شرکت ایلومینا که برای بررسی وضعیت متیلاسیون طراحی شده اند، آن است که پروب طراحی شده در سطح یک بید توانایی شناسایی سیتوزین متیله و غیر متیله ۷۲ درصد از لوکوس های ژنی را دارا می باشد. بنابراین، مابقی لوکوس ها با دو نوع بید مورد بررسی قرار می گیرند به طوریکه یک بید توانایی شناسایی قطعه DNA واجد سیتوزین متیله و بید دیگر به شناسایی سیتوزین غیر متیله می پردازد. در نتیجه عمده توالی های CpG به پروب نوع یک و مابقی به پروب های نوع دو متصل می شوند. اینکار سبب می شود توالی های بیشتری از پروب در هر چیپ بارگذاری شوند در حالی که اندازه چیپ افزایش نمی یابد. اما از معضلاتی که ایجاد می شود همپوشانی بخشی از سیگنال های ساطع شده از پروب نوع یک و دو می باشد که در این مرحله به صورت نرم افزاری اصلاح و تصحیح می گردد.
6. Adjusting batch/plate/chip confounders
اری های بررسی متیلاسیون DNA تا حد زیادی به نحوه آماده سازی هر Batch از یک نمونه حساس می باشد. این موارد مستقل از فاکتور های بیولوژیک نمونه بوده و عموما به شرایط و زمان آزمایش و آزمایشگاه بستگی دارد. بنابراین داپلیکیت های هر batch در این مرحله مورد بررسی قرار می گیرند. حتی بعضا علی رغم طراحی قابل قبول مطالعه، عواملی مداخله گر سبب اختلال در نتایج بدست امده می شود. برای مثال، حدی ازerror برای خطای چیپ های ساخته شده در نظر گرفته می شود که با نرمالیزه کردن نتایج قابل برطرف شدن نمی باشد. این موارد می تواند ناشی از emission بیش از حد و گاها بدلایل ناشناخته باشد.
7. آنالیزهای پایین دست
در این بخش دیتای خام بدست امده از میکرواری بوسیله نرم افزار های بیوانفورماتیک برای دستیابی به هدف مورد نظر (میزان متیلاسیون لکوس های ژنی و غیر ژنی) مورد بررسی قرار میگیرد.
i. وضعیت متیلاسیون
میزان یا درصد متیلاسیون در هر لوکوس قطعه DNA که وارد آزمایش شده براساس یک فرمول ریاضی محاسبه می شود
ii. Differential methylation/region-based analysis:
این آنالیز به بررسی ارتباط میان یک فنوتیپ دلخواه و متیلاسیون یک ناحیه CpG خاص می پردازد تا مناطقی که اصطلاحا به طور متفاوتی (مثلا در نمونه های بیمار و کنترل) متیله شده اند را مشخص کند.
iii. Clustering/profile analysis:
Clustering یا طبقه بندی به گروه بندی کردن نتایج میکرواری باز می گردد. به طوری که، اجزای هر گروه بیشترین شباهت را از نظر متیلاسیون با یکدیگر داشته و با سایر گروه ها تا حد امکان بیشترین اختلاف را دارند. این موضوع به ویژه در بررسی متیلاسیون بافت های سرطانی اهمیت می باید.
iv. آنالیز مسیر
بسیاری محققان از این آنالیز برای یافتن اهمیت نتیجه به دست آمده از متیلاسیون یک یا چند ژن در عملکرد خود ژن یا در مسیر های انتقال پیام استفاده می کنند.
8. Multiple testing correction
زمانی که آنالیز های فوق به نتایجی معتبری رسید، مرحله ای تحت عنوان multiple testing correction جهت کاهش احتمال وجود لوکوس هایی با نتایج مثبت کاذب انجام می گیرد. در واقع، با بکارگیری تست های آماری همچون Bonferroni correction میزان خطای مثبت کاذب از دیتای در دسترس کسر میگردد. این تست آماری با ایجاد داده های تصادفی مشابه دیتای به دست آمده، سپس آنالیز فوق را بارها تکرار می کند تا خطای آماری نتایج را سنجیده و موارد مثبت کاذب را گزارش کند.
9. Validation of significant hits
گام نهایی در پردازش اطلاعات میکرواری با هدف بررسی متیلاسیون DNA تایید نتایج به دست آمده، از طریق استفاده از چیپ های متفاوت و یا حتی روشی به جز میکرواری، می باشد. به ویژه در این میان پایروسکوئنسینگ به عنوان gold standard توصیه شده است.
پایگاه داده ها و ذخیره سازی داده
انبوهی از اطلاعات متیلاسیون سلول ها و بافت های مشتق از افراد سالم و بیمار جمع آوری و برای دسترسی ساده تر محققان در پایگاه های داده ذخیره می گردد. از جمله این موارد می توان به The Cancer Genome Atlas project (TCGA)، Gene Expression Omnibus (GEO)، iMETHYL، MethBank، DiseaseMeth، MethyCancer، MethDB، NGSmethDB، PubMeth، و MENT اشاره داشت. iMETHYL دیتابیسی برای ژنومیکس، پروتئومیکس و سایر omics ها می باشد که حاوی داده های حاصل از متیلاسیون DNA کل ژنوم به ویژه در مورد سلول های ایمنی می باشد. MethBank بر متیلاسیون DNA گونه های مختلف تمرکز داشته و به صورت دوره ای به روز رسانی می گردد. DiseaseMeth محتوی مجموعه دیتای متیلاسیون ۸۸ بیماری انسانی ست که مطالعات آنها گاهی بر ناحیه خاصی از DNA تمرکز داشته و گاهی به صورت توالی یابی کل ژنوم صورت گرفته است. MethyCancer محتوی دیتای ژنتیکی و ژنومیک مربوط به متیلاسیون سلول های سرطانی می باشد و مجموعه دیتاهای موجود در آن از طریق Cancer Epigenome Project کشور چین استخراج شده است. MethDB از جمله جالب ترین پایگاه داده های مرتبط با متیلاسیون DNA می باشد که طی پاسخ سلول به تغییرات محیط اطراف به دست آمده است. اطلاعات حاصل از متیلاسیون متفاوت سیتوزین در یک ناحیه خاص در شامپانزه و موش به صورت تجمیعی و یکپارچه در دیتابیس NGSMethDB گردآوری شده است. PubMeth نیز حاوی اطلاعات متیلاسیون به دست آمده از مقالات علمی منتظر شده می باشد. MENT نیز از ابتدایی ترین پایگاه های داده برای بررسی متیلاسیون و بیان ژن های مختلف در بافت های توموری می باشد. جالب آنکه، دیتابیس MethHC از گسترده ترین پایگاه های حال حاضر می باشد که بیش از ۵۰ هزار داده میکرواری و توالی یابی را از TCGA و GEO استخراج و به صورت یکپارچه در دسترس قرار داده است. در این دیتابیس همچنین شواهد علمی متیلاسیون بیش از ۷ هزار مقاله علمی استخراج و در محیطی ساده به صورت قابل نمایش در آمده است.
کاربردهای روش
همانطور که گفته شد بررسی متیلاسیون DNA به روش میکرواری محققان را قادر می سازد تا با درک وقایع اپی ژنتیک که به کنترل ژن های کلیدی فرآیند های زیستی می پردازند، امکان کنترل مسیرهای انتقال پیام، تکثیر و تمایز سلول را داشته باشند. بدین ترتیب علاوه بر آن که اثر اپی ژنتیک در بروز بسیاری بیماری های مشخص می شود، شاید راهکار درمانی قابل استفاده ای نیز برای بیماران قابل دستیابی باشد. همچنین، اگر الگوی قابل اطمینانی از تغییرات اپی ژنتیک ترسیم شود، حساسیت اپی ژنتیکی افراد برای ابتلا بسیاری بیماری ها قابل توضیح و پیش بینی خواهد بود.
روش های مبتنی بر سکوئنسینگ کل ژنوم
تاریخچه
پروسه اولین استخراج DNA توسط Friedrich Mietscher در سال ۱۸۶۹ تا کشف روش های توسعه توالی یابی ژنوم نتیجه تلاش های بی وقفه جوامع مختلف علمی در جهان می باشد. پس از آنکه Watson، Crick وFranklin موفق به کشف ساختار DNA در ۱۹۶۵ شدند، تلاش ها برای توالی یابیDNA آغاز شد. در سال ۱۹۷۲، Walter Fiers موفق شد اولین توالی یابی تاریخ را به خود ثبت کند. به طور خلاصه در این روش، ریبونوکلئاز T1 جهت هیدرولیز محل های خاصی از رونوشت یکی از ژن های کد کننده پوشش پروتئینی باکتریوفاژ MS2 مورد استفاده قرار گرفت و قطعات الیگونوکلئوتیدی با روش الکتروفورز و کروماتوگرافی جداسازی شد. با توجه به دسترسی به توالی آمینواسیدی پروتئین یاد شده نهایتا توالی ژن مذکور مشخص و ثبت گردید. اما جهش اصلی در دنیای توالی یابی ژنوم به نام Fredrick Sanger در سال ۱۹۷۷ ثبت شده است. روش او مبتنی بر توالی یابی قطعات کوچک درحال پلیمریزه شدن DNA می باشد و تحت عنوان روش اختتام زنجیره خوانده می شود. در این فرآیند، عمده نوکلئوتیدهای مورد استفاده جهت تکثیر DNA الگو از نوع دئوکسی نوکلئوتید تری فسفات (dNTPها) می باشد ولی بخشی از نوکلئوتیدها قادر به تشکیل باند فسفو دی استر با نوکلئوتید بعدی نبوده (ddNTPها) و سبب خاتمه فرایند پلیمریزاسیون می شوند. بعدها این روش توسعه یافت و جهت تسهیل کار از نوکلئوتیدهای فلوئورسنت و رادیواکتیو استفاده شد. بدین ترتیب قطعاتی از DNA با طول متفاوت تشکیل می شود که پس از الکتروفورز براساس نوع نوکلئوتیدی که در فرآیند سنتز DNA استفاده شده (دئوکسی آدنین، گوانین، سیتوزین، تیمیدین یا انواع دی دئوکسی آن ها) به راحتی می توان به توالی دقیق رشته الگو پی برد.
همزمان، Maxam و Gilbert پایه گذار روش نوینی از توالی یابی ژنوم شدند که اساس آن بر مبنای تغییرات شیمیایی توالی DNA می باشد و نیازی به پلیمریزاسیون الگو به کمک آنزیم DNA polymerase نمی باشد. در این روش که با عنوان توالی یابی شیمیایی نیز خوانده می شود، DNA نشاندار شده با 32P رادیواکتیو در مواجهه با مواد شیمیایی خاصی (مثل هیدرازین، دی متیلی سولفات و …) در محل آخرین نوکلئوتید دچار شکست شده و سپس همانند روش توالی یابی سنگر مورد الکتروفورز و آنالیز قرار می گیرد. این دو موفقیت، هم اکنون به عنوان اولین نسل روش های توالی یابی و یکی از دقیق ترین انواع آن می باشد. ولی فرایند انجام توالی یابی خودکار نبوده و وقت گیر می باشد. هم چنین این دو روش تنها برای توالی یابی قطعات DNA با حداکثر طول ۸۰۰ الی ۱۰۰۰ نوکلئوتید مورد استفاده قرار می گیرد.
اولین تکنولوژی از نسل دوم روش های توالی یابی مبتنی بر luminometry بوده و تحت عنوان pyrosequencing شناخته می شود. به بیان دقیق تر، نور ساطع شده از واکنش پیروفسفات حین پلیمریزاسیون DNA به واسطه آشکارسازهایی شناسایی و ثبت می گردد. در این روش نیز توالی هایی با طول حداکثر ۴۰۰ نوکلئوتید قابل توالی یابی می باشند اما سرعت کار به طور قابل توجهی بالا بوده و فرآیند به صورت خودکار قابل انجام است. با پیشرفت تکنولوژی، فناوری های توالی یابی جدید ظهور کرده و پلتفرم قدرتمندی برای تعیین توالی اگزوم فراهم کرد. انقلاب این ابزار ها زمانی رخ داد که دستگاه های مبتنی بر این تکنولوژی امکان توالی یابی همزمان کل ژنوم را به طور همزمان برای چندین نمونه داشتند به طوری که حداقل میزان خطا در خوانش توالی صورت بگیرد. این سیستم ها عمدتا توسط شرکت هایی همچون Illumina، Ion torrent وoxford nanaopore توسعه یافت و هم اکنون به عنوان ابزار اصلی برای تایید وقایع مبتنی بر تغییرات توالی (تغییرات ژنتیک) و اپی ژنتیک مورد استفاده قرار می گیرد. در بخش های دیگر با توضیحات جامع تر، فرآیند استفاده از این تکنولوژی در بررسی متیلاسیون DNA شرح داده خواهد شد.
تکنیک های مبتنی بر تیمار بی سولفیت
تا مدت ها بررسی متیلاسیون ها از طریق اندونوکلئازهای اختصاصی متیلاسیون انجام می گرفت. اساس این روش ها بر پایه ناتوانی آنزیم های محدودکنندهی حساس به متیلاسیون در برش سیتوزینهای متیله شده موجود در جایگاه تشخیص میباشند. مشکل اساسی که در رابطه با اندونوکلئازهای اختصاصی متیلاسیون وجود دارد این است که تنها سیتوزین هایی می توانند مورد بررسی قرار بگیرند که بتوانند توسط اندونوکلئازهای اختصاصی متیلاسیون موجود، تشخیص داده شوند به عنوان مثال،HpaII (CCGG) وHhaI (GCGC) . به علاوه، استفاده از این متدها مناسب آنالیزهای متیلاسیونی که روی کل ژنوم انجام میگیرد و نیز تکنیک های کشف نشانگرها است، اما در بررسی وضعیت متیلاسیون جایگاههای CpG اختصاصی چندان مفید عمل نمیکند. هم چنین، این تکنیک ها علیرغم سادگی، سرعت و حساسیت بالا، نسبت به روش های سکوئنسینگ حاضرنیازمند مقادیر بالایی از DNA با کیفیت هستند.
امروزه بسیاری از روش های موجود برای مطالعه تغییرات وضعیت متیلاسیون پروموترهای حاوی جزیره CpG وابسته به ایجاد تغییر در DNA تحت تاثیر نمک سدیم بیسولفیت می باشند. سپس تکثیر توسط PCR انجام میگیرد و امکان تمایز آللهای متیله شده و متیله نشده از یکدیگر فراهم میگردد. استفاده از سدیم بی سولفیت منجر به فیکس شدن الگوی متیلاسیون و برطرف کردن محدودیت های روش آنزیم محدود کننده می شود. تیمار DNA با بی سولفیت سبب میشود که بازهای غیر متیله سیتوزین گروه آمین خود را از دست بدهد و به باز یوراسیل تبدیل شود که همانند باز تیمین مکمل باز آدنین میباشد. بنابراین، باز سیتوزین بدون متیله به باز T تبدیل خواهد شد. اما در صورتیکه باز سیتوزین به صورت متیله باشد تیمار بیسولفیت اثری بر روی این باز نخواهد داشت. بنابراین به وسیله تیمار DNA به وسیله بی سولفیت و بررسی تبدیل باز C به باز T میتوان به متیله بودن و یا غیر متیله بودن بازهای C پی برد. پس از تیمار رشتههای DNA با بی سولفیت، تغییر بازهای C به Tبا روش PCR بررسی می شوند. اساس روش های ذکر شده، اصلاح شیمیایی سیتوزین در DNA تک رشته ای است. بنابراین مرحله دناتوراسیون DNA طی تیمار با بی سولفیت حائز اهمیت است. وجود یکی از رشته های DNA در ارزیابی های بیوانفورماتیکی برای عملکرد تکنیک های پایین دست مانند تکثیر، تعیین توالی و میکرواری لازم است. به طور کلی، کیفیت (نحوه استخراج) و کمیت (< 2µg) DNA تخلیص شده و PH واکنش برای استفاده از محلول بی سولفیت از عوامل حیاتی به شمار می روند. درمان ناقص بی سولفیت مشکل اصلی در روش های مبتنی بر بی سولفیت است که Zymo و همکارانش برای رفع این مسئله یک کیت تحقیقاتی تجاری بی سولفیت ارائه کرده اند.
به طور کلی روش های بررسی متیلاسیون DNA در سه بخش اصلی شامل
1) آماده سازی
2 تیمار DNA با بی سولفیت،
3) شناسایی مناطق متیله شده (PCR، تعیین توالی یا هضم آنزیمی)
طبقه بندی شده است. چندین عامل مهم متدولوژیکی در این تکنیک از قبیل مقیاس مطالعه (توان بالا، متوسط یا کم)، مقدار DNA آغازین، هزینه و زمان، حساسیت و دقت آزمایش و روش های کیفی یا کمّی وجود دارند. از این رو، چالش های رایج و نکات فنّی برای هر یک از روش ها مانند PCR، توالی یابی و واکنش های هضم آنزیمی وجود دارند که باید به منظور بهینه سازی واکنش های اختصاصی PCR، اجتناب از هضم آنزیمی ناقص یا غیر اختصاصی، مقرون به صرفه بودن و عیب یابی کلونینگ و توالی یابی مورد توجه قرار گیرند. ملاحظات دقیق برای بهینه سازی وضعیت دناتوره شدن DNA، تیمار بی سولفیت، شرایط انکوباسیون و ارزیابی DNA اصلاح شده پس از تیمار با بی سولفیت بسیار مهم است. پس از آماده سازی DNA، تیمار با بی سولفیت گام بعدی است. مطالعات کشت سلولی، آماده سازی های بیوپسی و مایعات بدن نیازمند جداسازی سلول ها و استخراج و تخلیص DNA می باشد.
متداول ترین روش هایی که برای بررسی متیلاسیون بر اساس تکنیک PCR استفاده می شوند عبارتند از:
(1) تکنیک PCR توالی یابی بی سولفیت
(2) تکنیک PCRاختصاصی متیلاسیون
(3) تکنیک ریل تایم PCR حساس به متیلاسیون
(4) تکنیک آنالیز منحنی ذوب با قدرت تفکیک بالا
(5) تکنیک پایروسکوئنسینگ DNA بی سولفیته
(6) تکنیک آنالیز تلفیقی روش برش و روش بی سولفیت
(7) تکنیک Reduced-representation bisulfite sequencing
(1) تکنیک توالی یابی بی سولفیت
این روش که توسط Frommer و همکارانش در سال 1992 توصیف شد، یک منطقه خاص را توالی یابی می کند تا اطلاعات کمّی در مورد محتوای C یا T در یک CpG خاص به دست آورد. این روش کلاسیک شامل مراحل تیمار با بی سولفیت، تقویت PCR و توالی یابی است. در روش BSPابتدا DNA استخراج شده از نمونه ها به وسیله بی سولفیت تیمار میگردد و سپس برای دو طرف جزیره CpG پرایمر طراحی شده و سپس قطعه تکثیر شده تعیین توالی میگردد. محصول یا به صورت مستقیم یا پس از کلون کردن آن در یک وکتور مناسب به وسیله توالی یابی سنگر توالی یابی می شود. دستور العمل های طراحی پرایمر BSP در ابتدا در سال 1994 توسط Clark و همکارانش منتشر شد. در این روش پرایمرها فاقد دی نوکلئوتیدهای CpG است. توالی پرایمر فوروارد بخشی از توالی قطعه مورد بررسی می باشد و با آن هم جهت است، اما توالی پرایمر معکوس به صورت reverse-complement با توالی قطعه مورد نظر می باشد. تمامی این مراحل می توانند به کمک نرم افزارهایی مانند METHYL PRIMER EXPRESS به انجام برسند. با مقایسه توالی به دست آمده با توالی مرجع می توان وضعیت متیلاسیون در ناحیه CpG را تفسیر کرد. وجود یک پیک C نشان دهنده وجود 5-متیل سیتوزین (5mC) در ژنوم است. تشخیص ژنومی 5mC به وسیله روش BSP به عنوان روش استاندارد طلایی برای تشخیص متیلاسیون DNA در نظر گرفته می شود.
مزایای روش:
1) تشخیص همزمان متیلاسیون در چندین سایت CpG
2) آنالیز کمّی متیلاسیون در امپلیکون
3) نیاز به یک جفت پرایمر
معایب روش:
1) عدم وجود اطلاعات در سطح تک مولکول
2) وجود نویزهای قابل توجه در نتایج توالی
آنالیز داده:
کمّی سازی سطح متیلاسیون با مقایسه نسبی ارتفاع پیک های سیتوزین و تیمین در هر سایت CpG تعیین می شود. پیک تیمین به عنوان یک CpG غیر متیله تفسیر می شود و پیک سیتوزین یک CpG متیله شده را نشان می دهد. تحلیل داده های خام حاصل از BSP اغلب دشوار است، بدین منظور از الگوریتم های ESME، BiQ Analyzer و BISMA برای تفسیر داده ها کمک گرفته می شود.
کاربرد روش:
این تکنیک برای تعیین پروفایل متیلاسیون کروموزوم های 6، 20 و 22 و هم چنین برای تشخیص متیلاسیون ژن های ناهنجار مانند سرطان استفاده می گردد.
(2) تکنیک PCR اختصاصی متیلاسیون
معرفی تکنیک متیلاسیون DNA اختصاصی توسط Herman و همکارانش در سال 1996 زمینه ساز پیشرفت های زیادی در پژوهش های متیلاسیون DNA گردید. MSP تجزیه و تحلیل کیفی و حساس هایپرمتیلاسیون پروموتر را در جزایر CpG در رده های سلولی و در نمونه های بالینی امکان پذیر می کند. در MSP، DNA تیمار شده با بی سولفیت با استفاده از مجموعه پرایمرهای اختصاصی تکثیر می یابد و نیازی به آنزیم محدود کننده حساس به متیلاسیون نمی باشد. MSP یک روش سریع است که برای تعیین پروفایل همزمان چندین نمونه استفاده می شود.
روش MSP نیازمند دو جفت پرایمر می باشد، یک جفت پرایمر فوروارد و معکوس اختصاصی برای DNA تیمار شده با بی سولفیت در حالت سیتوزین های متیله شده و یک جفت پرایمر فوروارد و معکوس اختصاصی برای DNA تیمار شده با بی سولفیت از نوع غیرمتیله. بنابراین برای هر نمونه دو واکنش PCR با هر جفت پرایمر انجام می شود و متیلاسیون نسبی بر اساس تفاوت مقادیر Ct آن ها محاسبه می گردد. تفسیر نتایج MSP آسان است؛ تکثیر با جفت پرایمرهای متیله، متیلاسیون جایگاه های CpG را نشان می دهد و تکثیر با جفت پرایمرهای غیر متیله، بیانگر عدم متیلاسیون در جایگاه های پرایمر میباشد. یک احتمال دیگر هم وجود دارد و آن هم این است که توالی توسط هر دو جفت پرایمر متیله و غیر متیله تکثیر شود که این بیانگر متیلاسیون جزئی این توالی می باشد. پرایمرهای روش MSP باید طولانی باشند (معمولاً بین 20 تا 30 باز) و اندازه آمپلیكون باید كوتاه باشد (بین 100 تا 300 جفت باز). بر اساس چندین گزارش، مناطق غنی از CG طول محصولات PCR را محدود می کند، بنابراین؛ طول بهینه باید حداکثر تا 400 bp باشد. هم چنین لازم است پرایمرها برای داشتن اختصاصیت بالا حداقل یک یا دو CpG در انتهای 3’ داشته باشند. در نهایت، وضعیت متیلاسیون DNA هدف به وسیله تصویربرداری ژل الکتروفورز مشخص می شود. قابل ذکر است که این روش تنها سیتوزین های واقع در محدوده پرایمر متیلاسیون را ارزیابی می کند. علاوه بر این، ناهمگونی متیلاسیون یک موضوع دیگر در تکثیر است که به فقدان الگوی DNA یکسان یا ناهمگونی متیلاسیون در DNA سلول ها اشاره دارد. یک نگرانی دیگر، وجود دو رشته DNA غیر مکمل پس از انکوباسیون بی سولفیت در واکنش PCR است که یکی از آنها به عنوان الگو است. پرایمرها نباید در این روش به رشته DNA نامطلوب متصل شوند. به نظر می رسد که در ابتدا پرایمر معکوس متصل شده و تکثیر می یابد، و سپس پرایمر فوروارد طبق جهت 5′-3’ تکثیر می شود.
آنالیز داده:
MSP در مقایسه با BSP یک رویکرد کیفی است. در نتیجه، برای تجزیه و تحلیل نیمه کمّی، می توان از نرم افزار ImageJ برای محاسبه شدت باند و مقایسه شدت ها با شدت استاندارد برای به دست آوردن درصد نسبی متیلاسیون استفاده کرد. با این حال، ساده ترین راه برای تعیین کمّیت MSP استفاده از PCR کمّی خاص متیلاسیون در زمان واقعی (qMSP) است. با استفاده از این روش، سطح نسبی متیلاسیون ژن مورد نظر در نمونه به صورت درصد محاسبه می شود. درصد متیلاسیون هر ژن برابر است با نسبت مقدار متوسط DNA ژن متیله شده به مقدار متوسط DNA ژن مرجع داخلی که در 100 ضرب می شود. از آن جا که طراحی پرایمر حائز اهمیت بالایی است، نرم افزارهای اختصاصی به این منظور طراحی شده است. برنامه طراحی پرایمرهای اختصاصی متیلاسیون را می توان در سایت urogene یافت. وجود تنها یک باند خاص از واکنش با پرایمرهای های اختصاصی متیله شده یا عدم وجود هر نوع آمپلیکون، ممکن است مورد مثبت کاذب یا منفی کاذب باشد. بنابراین، برای پیشگیری از این موارد، روش MSP باید دارای یک کنترل مثبت برای واکنش پرایمر غیر متیله شده، یک کنترل مثبت ( نمونه DNA که در in vitro متیله شده) و یک کنترل منفی فاقد DNA برای واکنش PCR باشد.
مزایای روش:
1) نیازمند مقادیر کمتری از DNA
2) تفسیر نتایج بسیار آسان
3) حساسیت بالا (قدرت تشخیص یک آلل متیله شده در ازای 100 آلل متیله نشده)
4) مقرون به صرفه بودن
5) عدم نیاز به کلونینگ و توالی یابی
محدودیت های روش:
1) این تکنیک روشی کیفی است و تنها حضور یا عدم حضور متیلاسیون در ناحیه مورد نظر را تشخیص می دهد ولی قادر به آنالیز کمّی متیلاسیون در نمونه الگو نمی باشد.
2) احتمال وجود نتایج مثبت کاذب یا منفی کاذب
کاربردهای بالینی:
1) بررسی میزان پاسخ بیماران سرطانی به داروهای آلکیله کننده
2) بررسی هایپرمتیلاسیون و غیر فعال شدن ژن های سرکوب کننده تومور
3) بررسی بیماران دارای نقص در متیلاسیون DNA
(3) Real-time PCR حساس به متیلاسیون
تکنیک ریل تایم PCR حساس به متیلاسیون مبتنی بر Real-time PCR و تیمار بی سولفیت که برای تشخیص کیفیت و کمّیت متیلاسیون توسعه یافته است.Eads و همکارانش فناوری TaqMan را برای تعیین وضعیت متیلاسیون DNA در مناطق ژنومی خاص معرفی کردند و آن را MethyLight نامیدند. در پروسه MethyLight، از دو پروب FAM به عنوان گزارشگر و TAMRA به عنوان خاموش کننده برای افزایش اختصاصیت متیلاسیون ناحیه مورد نظر استفاده می شود. نسبت متعادلی از توالی های تکثیر شده (متیله و غیر متیله) برای کسب تصاویر مناسبی از الگوهای متیلاسیون ضروری است. سطح متیلاسیون به عنوان نسبت اندازه گیری های حاصل از ژن هدف و ژن مرجع تعریف شده است. دستاوردهای پیشرفته تر پروب های TaqMan شامل تولید آمپلیکون فلوئورسنت حساس به متیلاسیون ((MS-FLAG و متیل سنگین است. در MS-FLAG، یک جفت پرایمر خاص برای اتصال به CpG متیله شده منطقه هدف طراحی شده است. پرایمرها حاوی الیگونوکلئوتید در انتهای 5’ با یک خاموش کننده و گزارش گر هستند که توسط اندونوکلئاز PspGI جدا شده است. در طول تکثیر، آنزیم PspGI نقاط تشخیصی دو رشته را قطع می کند و خاموش کننده برای تولید فلورسانس رها می شود. منطقه غیر متیله فاقد محصول PCR است. در روش متیل سنگین، مسدود کننده های الیگونوکلئوتید به CpGهای غیر متیله در واکنش PCR متصل می شوند. بنابراین، پروب ها قادر به اتصال به CpGهای غیر متیله نبوده و تکثیر نمی شوند، در حالی که پروب ها به CpG متیله متصل شده و در طول تکثیر نور فلوئورسنت آزاد می کنند. کیفیت و کمیت وضعیت متیلاسیون، تمایز توالی های متیله و غیر متیله و ناهمگونی متیلاسیون از طریق منحنی های ذوب و تکثیر ارزیابی می شوند. اگرچه Real-time PCR تشخیص متیلاسیون را تسهیل می کند اما برخی از مشکلات ناشی از ناهمگونی الگو و تکثیر الگوی غیر متیله هم چنان باقی است. MethyLight برای شناسایی مارکرهای پیش آگهی در سرطان سینه انسان و دهانه رحم استفاده شده است.
(4) تکنیک آنالیز منحنی ذوب با قدرت تفکیک بالا (HRM)
HRM یکی دیگر از روش های کمّی مبتنی بر تیمار بی سولفیت و Real-time PCR است که در آن آنالیز کمّی منحنی های ذوب قطعات DNA پس از تکثیر PCR انجام می شود. این روش برای اولین بار برای بررسی پلی مورفیسم های تک نوکلئوتیدی (SNP) DNA معرفی شد. در روش HRM از رنگ های فلوئورسنت مختلفی استفاده می گردد، که توانایی اتصال به DNA دو رشته ای را دارند و در غلظت بالا سبب مهار PCR نمی شوند که از جمله آنها به رنگ های SYTO 9 Green، Eva Green،SYBR Green اشاره کرد. به منظور طراحی پرایمرهای اختصاصی برای توالی های بی سولفیت می توان از نرم افزار Gene Runner استفاده کرد و پرایمر طراحی شده را در پایگاه داده BiSearch چک نمود. در بررسی وضعیت متیلاسیون، رشتهی متیله و غیر متیله با توجه به اختلاف توالی که توسط تیمار با بی سولفیت در آنها اعمال می گردد، دمای ذوب (منحنی ذوب) متفاوتی دارند. DNA ای که در ابتدا متیله شده بود، سیتوزین ها را حفظ می کند و دمای ذوب بالاتری نسبت به دمای غیر متیله شده دارد. در این روش برای کنترل وضعیت متیلاسیون منطقه مورد نظر DNA، کنترل تجاری متیله و غیر متیله وجود دارد. منحنی متیلاسیون نمونه ها در مقایسه با کنترل DNA ارزیابی می شود و به صورت درصد متیلاسیون محاسبه می شود. سوگیری در انتخاب ناحیه متیله و غیر متیله به عنوان الگو یک مشکل دیگری است که تغییرات جزئی در طراحی پرایمر آن را بهینه می کند. اشکال این روش ضرورت دست یابی به یک محصول PCR خالص است که در بسیاری از موارد مشکل است. با این وجود، اگر یک محصول PCR خالص به دست آید، حتی تفاوت های کوچک (5 تا 10%) در متیلاسیون DNA قابل تشخیص است. جذابیت استفاده از تکنیک HRM در سهولت، دقت و سرعت عمل آن نسبت به دیگر روش های تحلیل وضعیت متیلاسیون DNA است.
آنالیز داده:
میزان شدت متیلاسیون از روش مقايسه ای به کمک فرمول 2–∆∆Ct به محاسبه می شود. برای ارزیابی کمّی درصد متیلاسیون، چنانچه درصد ژن مورد نظر از کنترل بالاتر باشد، به عنوان نمونه متیله شده در نظر گرفته می شود.
(5) تکنیک پایروسکوئنسینگ DNA بی سولفیته
دکتر رونقی و همکارانش در سال 1998 طراحان پیشگام در تعیین پایروسکوئنسینگ بودند. این تکنیک در ابتدا به منظور بررسی SNPها مورد استفاده قرار گرفت. به منظور استفاده در توالی یابی، واکنشهای پایروسکوئنسینگ به صورت پیدرپی انجام میگیرند تا توالی DNA را در حین سنتز آن ثبت نمایند. DNA با استفاده از PCR تکثیر یافته و با پرایمر بیوتینیله شده نشاندار می گردد. تفاوت اصلی این روش با روشهای موجود استفاده از dNTP های دارای برچسب رادیواکتیو یا فلوئورسنت است. این روش بر اساس تشخیص نورسنجی از انتشار پیروفسفات (دو گروه فسفات به هم پیوسته) به دنبال اضافه شدن نوکلئوتیدها به زنجیره DNA در طول همانندسازی به دست می آید. این روش شامل فرآیندی دو آنزیمی است: در واکنش اول پیروفسفات به وسیله آنزیم ATP سولفوریلاز، به ATP تبدیل میشود و سپس ATP تولید شده به عنوان سوبسترا برای آنزیم لوسیفراز مصرف شده و لوسیفراز تولید نور متناسب با مقدار پیروفسفات میکند. نور ساطع شده توسط دوربین های متصل به صفحه سیلیکونی ثبت و به صورت پیک در یک گراف نمایان می شود. تبدیل بازهای C به T در DNA بی سولفیته می تواند به صورت کمّی سنجیده شود. در پایروسکوئنسینگ نسخه های متعددی از DNA در یک واکنش تکثیر می شود و ارزیابی متیلاسیون را تسهیل می کند. این تکنیک قادر است تفاوت های کوچک در متیلاسیون حتی تا 5 درصد را تشخیص دهد و یک رویکرد خوب برای نمونه های ناهمگون (به عنوان مثال سرطان) است، جایی که فقط بخشی از سلول ها دارای ژن متیله متفاوتی هستند. طول توالی خوانده شده در این روش در سال 2005 برابر bp 100-150 بود اما در سال 2009 شرکت Roche با انجام تغییراتی توانست طول توالی خوانده شده را به bp 1000 برساند.
مزایای روش:
1) دقت، انعطاف پذیری و پردازش به صورت موازی (آنالیز 96 نمونه به صورت همزمان)
2) عدم نیاز به پرایمر و نوکلئوتید نشاندار
3) عدم نیاز به الکتروفورز که فرآیندی طولانی و زمان بر است (امکان مشاهده نتایج در همان لحظه)
4) بدون نیاز به کلون کردن برای ازدیاد قطعه مورد نظر (تکثیر به کمک PCR)
معایب روش:
1) کاهش دقت شناسایی توالی های هموپلیمرها به خصوص بیشتر از 5 نوکلئوتید می باشد.
2) هزینه ی بالای توالی یابی با این روش در مقایسه با سایر روش های توالی یابی در مقیاس وسیع
کاربرد روش:
برای ارزیابی پروفایل متیلاسیون DNA در مطالعات متعدد سرطان، از جمله بدخیمی های زنان و زایمان، از روش پایروسکوئنسینگ استفاده شده است.
(6) آنالیز تلفیقی روش برش و روش بی سولفیت
برای اولین بار در سال 1997 روش COBRA جهت بررسی کمّی هایپرمتیلاسیون ابداع شد. اساسا این روش برای آنالیز مقادیر اندک DNA از نمونه های بافتی جدا شده از پارافین ایجاد شده است. از آن زمان تاکنون این روش کاربرد گسترده ای در تحقیقات سرطان و مطالعات اپی ژنتیک داشته است. طراحی پرایمر در این روش مشابه BSP میباشد با این تفاوت که در این روش تشخیص متیلاسیون توسط آنزیمهای محدودکننده صورت میپذیرد. برای دو طرف منطقه CpG یک جفت پرایمر طراحی میشود و بعد از تکثیر توالی توسط پرایمرها، محصولات آن با آنزیم محدودکننده تیمار میشود. در واقع بعد از تیمار DNA با بیسولفیت و انجامPCR ، محصولات آن تحت برش آنزیمی قرار میگیرند. از آنزیم هایی مانند TaqI ,BstUIو MluI برای برش استفاده می شود. با توجه به ردیف بازهای اولیه DNA این آنزیم بهگونهای انتخاب میشود که عمل برش را فقط در جزایر CpG دارای هایپرمتیلاسیون انجام دهد. میزان هایپرمتیلاسیون در DNA اولیه با میزان کمّی برش محصولات PCR متناسب است. اگر CpG درDNA اولیه فاقد هایپرمتیلاسیون باشد در اثر تیمار بیسولفیت سیتوزین تبدیل به تیمین شده و جایگاه برش آنزیم از بین می رود و عمل برش انجام نخواهد شد. اگر در DNA اولیه مخلوط آللهای متیله و غیرمتیله وجود داشته باشد در زمان هضم آنزیمی، میزان برشها وابسته به مقدار CpG های متیله دارد. سپس قطعات برش یافته توسط الکتروفورز ژل پلی اکریل آمید به یک باند بزرگ هضم نشده و چندین باند کوچک تر مربوط به قطعات هضم شده جدا می شوند. با غلظت سنجی باندها در ژل پلی اکریل آمید (با کمک نرم افزار ImageJ) میزان کمّی هایپرمتیلاسیون محاسبه میشود. کمّی سازی متیلاسیون از مقایسه نسبت بین محصولات PCR هضم شده و باقی مانده برای ارزیابی میزان سیتوزین های متیله و غیر متیله در منطقه مورد نظر DNA انجام می گیرد.
محدودیت های روش:
1) در صورتیکه برای توالی مورد بررسی آنزیم محدود کننده ای یافت نشود نمیتوان از این روش استفاده نمود.
2) هضم ناقص توسط آنزیم های محدود کننده پس از PCR، عدم وجود متیلاسیون DNA را نشان می دهد (در صورت برش با آنزیم حساس به متیلاسیون مانند HpaII)
3) در نمونه های پیچیده، ناهمگونی نوع سلول می تواند بررسی را دچار اشتباه کند زیرا DNA به درستی توالی یابی نمی شود. به ویژه، ناهمگونی در توالی سلول های مختلف نمونه (یعنی جمعیت سلول های مختلف درون تومور) که دچار جهش هایی در منطقه مورد نظر شده اند، مانند تغییر دی نوکلئوتید CG به CA یا CT، منجر به از دست دادن محل محدودیت می شود و باعث ایجاد یک منطقه ظاهرا متیله شده به دلیل عدم هضم می شود. در این صورت کمّی سازی سطوح متیلاسیون DNA در یک نمونه دچار تحریف می گردد.
کاربرد روش:
1) غربالگری تغییرات متیلاسیون DNA در پروموترهای ژن در مطالعات سرطان (به عنوان مثال، مطالعه متیلاسیون DNA در ژن HPP1 در سرطان کولورکتال)
2) شناسایی الگوهای تغییر یافته متیلاسیون در ژن های حک شده
3) توصیف الگوهای متیلاسیون در ژنوم در طی تکامل در پستانداران
4) تشخیص اختلال ژنتیکی سندرم Russell-Silver (هیپو متیلاسیون ژن H19 در 50% بیماران)
(7) تکنیک Reduced-representation bisulfite sequencing
یک روش کارآمد برای تجزیه و تحلیل پروفایل متیلاسیون در سطح ژنوم در سطح نوکلئوتیدی است. با توجه به هزینه زیاد و عمق تعیین توالی برای بررسی وضعیت متیلاسیون در کل ژنوم، Meissner و همکارانش این روش را در سال 2005 برای تعیین توالی 1% ژنوم توسعه دادند. در این روش مقدار نوکلئوتیدهای مورد نیاز کاهش یافته است. این محققان برای بررسی صحّت تکنیک، متیلوم سلول های بنیادی جنینی موش (ES ) را با سلول های ES فاقد DNA متیل ترانسفراز (Dnmt3a, 3b) و با سلول های ES دارای سطوح کاهش یافته Dnmt1 مقایسه کردند. آن ها از BglII به عنوان آنزیم محدود کننده غیر حساس به متیلاسیون برای هضم DNA ژنومی استفاده کردند. قطعات DNA به طول 500-600 جفت باز انتخاب و کلون شد تا توالی یابی شود. این محققان DNA را با استفاده از فناوری توالی یابی سنگر توالی یابی کردند. با گذشت سال ها، روش ها و پروتکل های RRBS بهبود یافته اند. اول اینکه به جای BglII، آنزیم MspI برای پوشش بهتر سایت های CpG استفاده شد. ثانیا با پیشرفت در فناوری های توالی یابی DNA، توالی یابی سنگر با تعیین توالی نسل بعدی (NGS) جایگزین شد. در تکنیک RRBS ترکیبی از آنزیم های محدود کننده و توالی یابی بیسولفیت برای بررسی الگوهای متیلاسیونی در نواحی غنی از CpG مشخص مورد استفاده قرار می گیرد. در این روش ابتدا DNA توسط آنزیم محدود کننده غیر حساس به متیله شدن (MspI) به قطعات کوتاه برش داده می شود. در انتهای این قطعات کوتاه، دی نوکلئوتیدهای CpG هستند. آنزیم MspI توالی های 5′-CCGG-3’ را شناسایی می کند. قطعات غنی از CpG قبل از تیمار با بی سولفیت و PCR انتخاب می گردند. این قطعات باید به طول 40 تا 220 bp باشند و در 85 درصد نواحی CpG حضور داشته باشند. سپس، در مرحله بعد تیمار بی سولفیت انجام می گیرد و سیتوزین های متیله شده از تبدیل بی سولفیت محافظت می شوند، بنابراین هنوز در آنالیز به عنوان سیتوزین خوانده می شوند. DNA تیمار شده با بی سولفیت قبل از تعیین توالی توسط PCR تقویت می شود. سپس، توالی یابی نسل بعدی بر روی توالی های تکثیر شده انجام می شود. رایج ترین گردش کار RRBS از پلتفرم Illumina NGS استفاده می کند. پس از اتمام تعیین توالی، آخرین مرحله در پروتکل RRBS تجزیه و تحلیل بیوانفورماتیکی است که شامل ترازبندی توالی و شناسایی متیلاسیون است. پروتکل های مبتنی بر RRBS مقرون به صرفه تر از توالی یابی بی سولفیت کل ژنوم WGBS هستند، زیرا این روش ها بر تقویت مناطق غنی از CpG در نزدیکی توالی تشخیص آنزیم محدود کننده تمرکز دارند.
مزایای روش:
1) پوشش دهی بالای نواحی جزایر CpG (CGI)
2) حساسیت بالا
3) مقرون به صرفه بودن در مقایسه با WGBS
4) نیاز به DNA کمتر (10-300 ng)
محدودیت های روش:
1) پوشش دهی پایین در عوامل تنظیمی دور و مناطق داخل ژنی
2) تخریب قابل توجه DNA پس از تیمار با بی سولفیت
3) محدود به مناطق نزدیک به سایت های شناسایی آنزیم ها
4) عدم افتراق بین 5MC و 5hmC
آنالیز داده:
فرآیند کلی تجزیه و تحلیل بیوانفورماتیکی داده های متیلاسیون DNA شامل پردازش داده ها، کمّی سازی سطح متیلاسیون DNA، تعیین پروفایل عمومی، شناسایی DMR (مناطق با متیلاسیون متفاوت) و تجسم متیلوم است. کنترل کیفیت داده ها با استفاده از نرم افزار FastQC انجام می شود.
ابزارهای آنالیز اطلاعات RRBS:
BS Seeker، Bismark و BSMAP ابزارهای مناسب برای خوانش ردیف های توالی یابی بیسولفیت هستند. BS Seeker ردیف یابی و فراخوانی متیلاسیون را انجام می دهد ولی نسبت متیلاسیون یا نمرات بتا را محاسبه نمی کند. الگوریتم Bicycle قادر به انجام تمام مراحل محاسباتی ضروری است و برای سیستم عامل های مختلف عمومیت دارد. Bicycleبه علت استفاده آسان تر و عملکرد بالا بهترین ابزار محاسباتی مورد پسند کاربران به خصوص دانشمندان بدون دانش محاسباتی می باشد. SMAP یک نمونه عالی پایپلاین است که تنها برای داده های RRBS مناسب است. MOABS نیز یکی از قدرتمندترین پایپلاین های خطی است که برای آنالیز داده های WGBS، RRBS و 5hmC کاربرد دارد و برای کاربران با تجربه و متخصص انتخاب مناسبی است. Genestack.com (https://genestack.com) یک پلتفرم آنلاین برای آنالیز انواع داده های متیلاسیون از جمله WGBS و RRBS است که در آن می توان داده های شخصی خود را بارگذاری و تحلیل کرد.
مشخصات تراز بندی را می توان با ابزارهایی مانند UCSC genome browser، WBSA، Integrative Genome Viewer و Methylation plotter ترسیم کرد که منجر به وضوح بیشتر در سطح یک باز در سراسر ژنوم می شود.
کاربرد بالینی:
1) تشخیص تغییر الگوهای متیلاسیون در تومورها نسبت به بافت طبیعی (همچون سرطان دهان، معده، کولورکتال، سینه و لنفوم)
2) تعیین الگوهای متیلاسیون در زمینه بیماری های اعصاب مانند هانتیگتون (HD ) و سندرم داون (DS )
3) تعیین تغییر الگوهای متیلاسیون DNA در پانکراس افراد دیابتی