
جدول محتوا
مقدمه
پیشرفت های اخیر در زمینه ژنتیک، قابلیت تعیین توالی DNA را افزایش داده است. براساس توسعه و رشد تکنولوژی های تعیین توالی نسل دوم (NGS)، استفاده از روش توالی یابی کل ژنوم (WGS) برای بررسی اساس ژنتیکی بیماری های مختلف بسیار رایج شده است. پلتفرم های مختلف تعیین توالی نسل دوم، میلیون ها قطعه ی DNA را به صورت موازی تعیین توالی می کنند و اجازه می دهند که توالی کل ژنوم در یک مرحله مشخص شود. علم بیوانفورماتیک توالی این قطعات را در مقایسه با ژنوم رفرانس، کنار هم قرار می دهد تا توالی کل ژنوم مشخص شود. رویکرد توالی یابی کل ژنوم در تحقیق، تشخیص و شناخت مکانیسم بیماری ها و توسعه هدف های درمانی مفید و پرکاربرد می باشد.
ژنوم حاوی اطلاعات کامل ژنتیکی یک ارگانیسم یا یک سلول است. این اطلاعات حاوی اسید های نوکلئیک تک یا دو رشته ایی هستند که به صورت توالی های خطی یا حلقوی ذخیره می شوند. برای تعیین دقیق این توالی ها، تکنولوژی های دقیق، با توان و سرعت بالا در تعیین توالی ژنوم توسعه پیدا کردند. دستگاه های تعیین توالی، توالی هایی تحت عنوان “خوانش ها” با سایز کوتاه را در سراسر ژنوم تولید می کنند. سکانس کامل ژنوم از همپوشانی این قطعات کوتاه تحت فرایند مونتاژ جدید یا مونتاژ از نو به دست می آیند. در پروژه های تعیین توالی از توالی هایی “رفرانس” مخصوص هر ارگانیسم به عنوان الگو جهت الاینمت یا همترازی خوانش ها استفاده می شود. توالی های ژنومی به دست آمده در شناخت و مطالعه عملکرد ژن ها و طراحی مطالعات دستکاری ژن ها مورد استفاده قرار می گیرد. در سال های اخیر، روش های جدید تعیین توالی، تعیین توالی نسل دوم (NGS)، منجر به شناخت سطح وسیع تری از ژنوم شده اند و تحت عنوان توالی یابی کل ژنوم (WGS) توسعه یافته اند. علاوه بر این، در دو دهه ی اخیر به دنبال توسعه توالی یابی کل ژنوم، روش های جدیدتر تعیین توالی نسل سوم به عنوان بازیگرهای قوی در ژنومیک پدید آمده اند. روش های تعیین توالی نسل سوم، با توانایی تولید توالی هایی به طول ده ها تا هزاران کیلوباز و صحت نزدیک به تکنولوژی های با خوانش کوتاه، قدرت خود را در حل برخی از مشکلات تعیین توالی در نواحی خاصی از ژنوم به اثبات رسانده اند. از جمله این موارد می توان به شناسایی واریانت های ساختاری در نواحی غیر قابل دسترس اشاره کرد. در این مطالعه، اصول و انواع روش های تعیین توالی ژنوم بررسی می شود.
تاریخچه
روش های تعیین توالی DNA در دهه های 1970 و 1980 با استفاده از روش تعیین توالی یابی ماکسیم گیلبرت و روش تعیین توالی سنگر شروع شد. اولین مولکول DNA که در سال 1975 به طور کامل توالی یابی شد ژنوم باکتریوفاژ phiX174 با 5386 نوکلئوتید بود. به تدریج تعیین توالی برای مولکول های بزرگ تر نیز انجام شد. تیم تحقیقاتی پروفسور سانگر توالی ژنوم میتوکندری انسان (16/6kb) را در سال 1981 و توالی ژنوم باکتریوفاژ لامبدا را در سال 1982 منتشر کردند.
در طول دهه 1990 روش های خودکار توالی یابی خاتمه زنجیره توسعه یافت و تعیین توالی ژنوم های بزرگ تر امکان پذیر شد. توالی اولین کروموزوم یوکاریوتی، یعنی کروموزوم شماره III مخمر ساکارومایسس سروزیه، در سال 1992 منتشر شد و در سال 1996 توالی یابی کل ژنوم مخمر تکمیل شد. در ادامه توالی یابی کل ژنوم ارگانیسم های مدل مانند کرم نماتود سی-الگانس، مگس دروزوفیلا ملانوگاستر انجام شد. در سال 1999 توالی کامل کروموزوم 22 انسانی، کوتاهترین کروموزوم اتوزوم انسانی، منتشر شد. همین طور توالی ژنوم موش های آزمایشگاهی Mus musculus و انسان به ترتیب در سال های 2002 و 2001 منتشر شدند. با توسعه روش های توالی یابی نسل جدید، توالی یابی ژنوم فرایندی معمول شده است و توالی کامل ژنوم گونه های مختلف یوکاریوتی و پروکاریوتی به کمک پروژه های زیادی که در حال انجام هستند، شناسایی شده اند. در سال 2008 توالی ژنوم اولین فرد مونث گزارش شد. علاوه بر این در سال 2010 اولین مونتاژ موفقیت آمیز مربوط به ژنوم پاندای غول پیکر تنها با استفاده از روش های نسل جدید گزارش شد.
اساس روش توالی یابی کل ژنوم
مروری بر توالی یابی کل ژنوم
ژنوم هر ارگانیسم حاوی تمام اطلاعات ژنتیکی آن است. فناوری توالی یابی ژنوم قادر است به طور جامع و دقیق کل ژنوم را تجزیه و تحلیل کرده و اطلاعات موجود در آن، پیچیدگی و تنوع ژنوم را آشکار می کند. ظهور فناوری توالی یابی ژنوم یک پیشرفت انقلابی در تمام زمینه های علوم زیستی است. توالی یابی کل ژنوم می تواند انواع تغییرات، از جمله انواع تک نوکلئوتیدی، درج/حذف ها، تغییرات تعداد کپی و انواع تغییرات ساختاری در مقیاس بزرگ را تشخیص دهد. بسته به اینکه ژنوم رفرانس وجود داشته باشد و یا نه، توالی کل ژنوم را می توان به دو نوع تعیین توالی مجدد و تعیین توالی جدید/از نو طبقه بندی کرد.
در دهه 1990 که روش های خودکار توالی یابی ژنوم توسعه یافتند، به دست آوردن اطلاعات درباره توالی، عامل محدود کننده ای در پروژه های توالی یابی ژنوم نبود. بلکه چالش اصلی کنار هم قرار دادن هزاران تا میلیون ها توالی کوتاه برای به دست آوردن یک توالی ژنوم بهم پیوسته بود. ساده ترین راه برای انجام این کار، روش تعیین توالی “شاتگان” بود که در آن ژنوم به طور تصادفی به قطعات کوتاه شکسته شده و این قطعات به طور جداگانه توالی یابی می شوند. توالی پیوسته ژنوم با بررسی همپوشانی بین توالی های کوتاه بدست می آید (شکل 1).
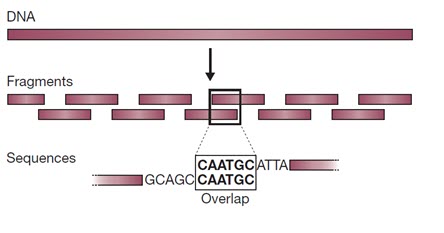
شکل 1. روش شاتگان برای مونتاژ توالی ها است. مولکول DNA به قطعات کوچکی شکسته می شود که هر یک از آنها توالی یابی می شوند. توالی اصلی با جستجوی همپوشانی بین توالی این قطعات فراهم می شود.
دو رویکرد کلاسیک برای تعیین توالی ژنوم های بزرگ
در اوایل دهه 80، با استفاده از روش شاتگان توالی کامل ژنوم فاژ لامبدا با موفقیت تعیین توالی شد و این روش با موفقیت در تعیین توالی DNA هایی با اندازه بزرگتر نظیر DNA ویروسی، DNA اندامک و توالی ژنوم باکتری استفاده شد. تعیین توالی شاتگان یک روش کلاسیک برای تعیین توالی کل ژنوم است. روش تعیین توالی شاتگان تعیین توالی در مقیاس بزرگ را برعهده دارد.
توالی یابی شاتگان روشی است که DNA را برای توالی یابی و مونتاژ مجدد به صورت تصادفی بخ قطعات کوچک می شکند. قطعات DNA نیز برای تکثیر و تعیین توالی بعدی در باکتری کلون می شوند. چون قطعات به طور تصادفی ایجاد شده اند، توالی های همپوشان وجود دارند که به مونتاژ مجدد مطابق ترتیب DNA اصلی کمک می کنند. این رویکرد در ابتدا در توالی یابی سنگر استفاده می شد اما اکنون در روش های توالی یابی نسل جدید که توالی یابی سریع و موازی کل ژنوم را انجام می دهند نیز استفاده می شود. این روش فقط برای “خوانش”های کوتاه مناسب است، به این معنی که توالی یابی برای قطعات DNA کوتاه تر که دوباره کنار هم قرار گیرند انجام می شود. روش توالی یابی شاتگان برای توالی یابی کل ژنوم عمدتا شامل دو نوع است: توالی شاتگان سلسله مراتبی (روش کلون به کلون) و دیگری توالی شاتگان کل ژنوم.
توالی یابی شاتگان سلسله مراتبی
توالی اولین ژنوم یوکاریوتی از جمله انسان، توسط اصلاحاتی در روش توالی یابی (با عنوان توالی یابی شاتگان سلسه مراتبی) انجام شد. این روش دارای یک مرحله قبل از توالی یابی است که در آن DNA ژنومیک به قطعاتی کوچکتر و معمولا به اندازه 300 کیلوباز شکسته می شود و سپس این قطعات در وکتورهایی با ظرفیت بالا مانند BAC کلون می شوند. سپس هر قطعه از DNA کلون شده به قطعات کوچکتر شکسته می شود و سپس توالی یابی انجام می شود و سپس با روش شاتگان مونتاژ می شوند. توالی هر قطعه کلون شده در موقعیت مناسب خود قرار می گیرد و توالی کلون های حاوی قطعات DNAی همپوشان شناسایی شده و به تدریج توالی ژنوم ساخته می شود (شکل 2).
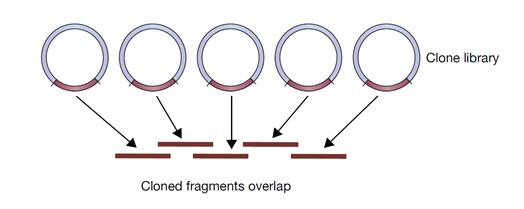
شکل 2. مجموعه ای از کلون ها که قطعات آنها با یکدیگر همپوشانی دارند.
تعیین توالی ژنوم با توالی یابی شاتگان
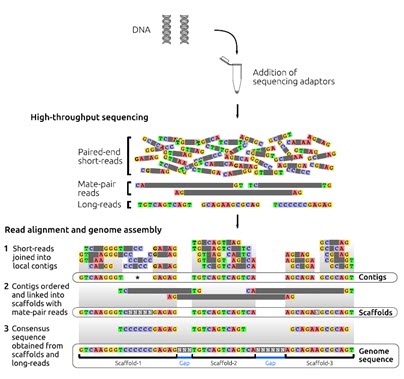
اغلب پرژه های ژنومی از استراتژی تعیین توالی شاتگان برای دستیابی به توالی ژنوم استفاده می کنند. تعیین توالی ژنوم به طور کلی شامل شش مرحله است: استخراج DNA ژنومی، تولید قطعات تصادفی DNA ژنومی، انتخاب اندازه قطعات تولید شده با استفاده از الکتروفورز، ساخت کتابخانه، تعیین توالی با انتهای جفت شده (تعیین توالی PE) و مونتاژ ژنوم است.
به صورت خلاصه، در مرحله اول، DNA ژنومیک به صورت تصادفی قطعه قطعه می شود. قطعات DNA با دو اندازه متفاوت شامل درج (insert) بلندتر (2.5-2کیلوبایت) و درج کوتاهتر (1.2-0.5کیلوبایت) با استفاده از ژل آگارز انتخاب می شوند. در حالی که درج های بلند در فاژ یا وکتورهای کاسمید کلون می شوند، درج های کوتاه در وکتورهای پلاسمید کلون می شوند. کتابخانه کلون قطعات درج کوتاه برای تعیین توالی از دو انتها استفاده می شود.
بعد از تعیین توالی با استفاده از الگوریتم های کامپیوتری قدرتمند، خوانش های به دست آمده برای تشکیل قطعات بهم پیوسته بلند DNA ترکیب و کنارهم قرار می گیرند (هر یک از این قطعات اصطلاحا کانتیگ نامیده می شود) که کل این فرایند به مونتاز از نو (denovo assembly) معروف است. برای مونتاژ صحیح، لازم است که همپوشانی کافی بین خوانش ها در هر موقعیتی از ژنوم وجود داشته باشد و به همین دلیل تعیین توالی با پوشش یا عمق خوانش بالا لازم است. به طور طبیعی خوانش های بلندتر همپوشانی بیشتری خواهند داشت که در نتیجه باعث کاهش عمق مورد نیاز خوانش ها می شود. معمولا قطعات بلندتر (چند صد جفت باز) از هر دو انتها توالی یابی می شوند (توالی یابی با انتهای جفت شده) تا خوانش ها در جهت و مکان صحیح در مونتاژ ژنومیک قرار گیرند. بعد از مونتاژ اولیه، کانتیگ ها به هم ملحق می شوند و دنباله ها یا قطعات بلندتری را به نام اسکافولد تشکیل می دهند. برای رسیدن به این هدف، کتابخانه هایی با قطعات بلند DNA از ژنوم (چند کلیو باز) تهیه می شود و نقاط انتهایی تعیین توالی می شوند. با توجه به نوع تکنولوژی و ویژگی های کتابخانه های تهیه شده، این کتابخانه ها اینگونه نامیده می شوند: paired-end، mate-pair یا jump. از همپوشانی و کنار هم قرار گرفتن کانتیگ ها، اسکافولد ها ایجاد می شوند. اندازه قطعات در کتابخانه، اطلاعاتی در مورد فاصله فیزیکی بین دو کانتیگ فراهم می کند و شکاف ایجاد شده با جفت بازهای غیرآگاهی دهنده پر می شوند. پر کردن شکاف ها با استفاده از خوانش های بلند باعث می شود که نواحی تکراری پوشش داده شود و به دستیابی اطلاعات بازها در این نواحی کمک می کند. با کنار هم قرار گرفتن کانتیگ ها اسکافولدهای بلندتر هم تشکیل می شوند و این اسکافولدها روی کروموزوم ها ملحق و کنار هم قرار می گیرند (شکل 3).
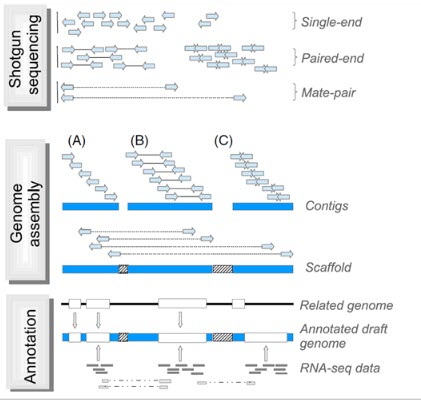
شکل 3. شمایی ساده از فرایند مونتاژ در تعیین توالی ژنوم. توالی یابی شاتگان: قطعات کوتاه DNA در موقعیت های تصادفی در سراسر ژنوم برای دستیابی به پوشش عمق خوانش ها توالی یابی می شوند. قطعات می توانند شامل تک خوانش ها (یا unpaired reads، به طول 50-1000bp) یا خوانش های با انتهای جفت شده (paired reads) (توجه داشته باشید که خوانش های با انتهای جفت می توانند با هم همپوشانی داشته باشند) و همراه با قطعات درج شده با اندازه های متنوع باشند. کتابخانه های mate-pair نواحی ژنومی بزرگتر را در بر می گیرند. مونتاژ ژنوم: (A) مونتاژکننده های جدید با استفاده از خوانش های کوتاه، اطلاعات توالی خوانش ها را در دنباله های پیوسته به نام کانتیگ قرار می دهند. (B) خوانش های با انتهای جفت شده اطلاعات بیشتری را برای خوانش ها ی یک کانتیگ فراهم می کنند. حاشیه نویسی: مدل های ژنی توسط الگوریتم های پیش بینی کننده به صورت in silico بدست می آیند. این مدل های ژنی با استفاده از اطلاعات ژنومی ارگانیسم های مرتبط با ژنوم مورد هدف (ژنومی که قرار است تعیین توالی شود) و با استفاده از داده های بیانی (از ارگانیسم مورد هدف در تعیین توالی) مانند RNA-seq فراهم می شوند. خوانش های برش داده شده از داده RNA-seq اطلاعات ارزشمندی از splice junctions و ایزوفرم های مختلف یک ژن فراهم می کنند.
مونتاژ خوانش های تعیین توالی
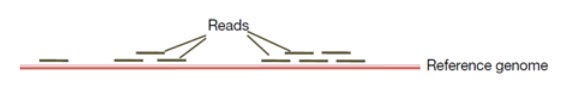
دو روش برای تعیین توالی ژنوم ذکر می شود: تعیین توالی ژنوم بر اساس مونتاژ جدید یا از نو و تعیین توالی ژنوم بر اساس مقایسه با ژنوم رفرانس. توالی یابی با روش مونتاژ جدید در ژنوم بسیاری از پروکاریوت ها از جمله هموفیلوس آنفولانزا انجام شده است. هرچند روش دیگری جهت مونتاژ کردن ژنوم وجود دارد که از توالی ژنوم رفرانس یک موجود برای مونتاژ توالی ژنوم مورد هدف استفاده می شود (شکل 4). در روش دوم، ژنوم رفرانس مربوط به گونه ای مرتبط یا سویه متفاوتی از همان گونه است، بدین ترتیب ژنوم توالی یابی شده با ژنوم رفرانس مقایسه می شود و در این صورت ژنوم رفرانس می تواند مونتاژ ژنوم جدیدی را هدایت کند. با استفاده از ژنوم رفرانس مونتاژ توالی ها بسیار سریع تر و دقیق تر انجام می شود.
تعیین توالی ژنوم بر اساس مونتاژ جدید
روش تعیین توالی ژنوم بر اساس مونتاژ جدید (De-novo assembly) توالی و مونتاژ یک ژنوم کامل برای اولین بار است. در این تعیین توالی ژنوم، DNA ژنومی با کیفیت بالا برای تهیه کتابخانه (library) قطعه قطعه می شوند و سپس توالی آداپتورها به این قطعات DNA اضافه می شوند. خوانش های کوتاه با انتهای جفت شده (100جفت باز) با استفاده از دستگاه های تعیین توالی با قدرت بالا، از کتابخانه هایی از قطعات DNA با اندازه های مختلف برای به حداکثر رساندن پوشش ژنوم به دست می آیند. خوانش های بلند (2-10 کیلوبایت) به مونتاژ نواحی تکراری کمک می کنند. شکل 5 تعیین توالی ژنوم را بر اساس مونتاژ جدید نشان می دهد.
تعیین توالی ژنوم بر اساس مقایسه با ژنوم رفرانس
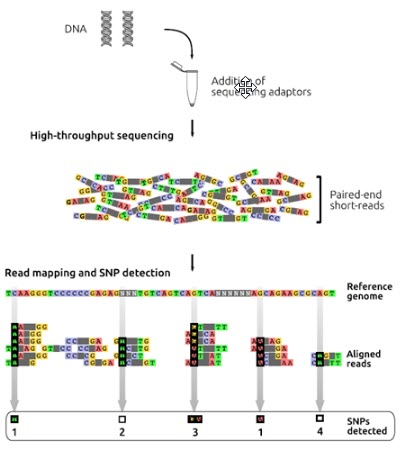
در این روش تعیین توالی ژنوم، توالی های ژنومی افراد مختلف با ژنوم رفرانس مقایسه یا الاین می شوند. DNA ژنومی با کیفیت بالا برای تهیه کتابخانه قطعه قطعه می شود و سپس به قطعات DNA، آداپتورهای تعیین توالی اضافه می شود و همچنین اندازه قطعه درج شده 500-350 جفت باز است. خوانش های کوتاه با انتهای جفت شده (100p) از کتابخانه DNA با استفاده از دستگاه های توالی یابی با توان بالا بدست می آیند. خوانش های کوتاه بر اساس شباهت توالی با ژنوم رفرانس الاین می شوند (شکل6). پلی مورفیسم های تک نوکلئوتیدی (SNP) زمانی تشخیص داده می شوند که یک باز خاص در موقعیتی از ژنوم رفرانس با باز مشاهده شده در خوانش ها متفاوت باشد. در بررسی SNP ها باید به پوشش ناهموار خوانش ها در برخی موقعیت ها و هم چنین مواردی ذیل توجه شود: (1) برخی تغییرات فقط در خوانش ها وجود دارند اما در ژنوم رفرانس وجود ندارند، (2) برخی از SNP ها ممکن است تشخیص داده نشوند زیرا در ژنوم رفرانس وجود ندارند، (3) برخی از SNP ها ممکن است در خوانش ها به صورت هتروزیگوت باشند (4) و برخی از آنها ممکن است در حین فیلترینگ در پروژه های تعیین توالی حذف شوند.
روش آزمایشگاهی توالی یابی کل ژنوم
نمونه برداری و حفظ نمونه
رعایت شیوه های مناسب درجمع آوری و حفظ نمونه ها جهت جلوگیری از آسیب DNA الزامی است.
استخراج DNA و کنترل کیفیت و کمیت آن
کیفیت DNA با استفاده از الکتروفورز ژل آگارز 0.8-1 درصد و لدر با وزن مولکولی 25-Kbp ارزیابی می شود. یک باند با وزن مولکولی بالا (~23 Kbp) DNA نشان دهنده یکپارچگی خوب DNA است. خلوص بالای DNA با نسبت جذب 260/280 نانومتر 1.8-2 ~ تأیید می شود. از قطعه قطعه شدن زیاد DNA باید اجتناب شود چرا که کمیت DNA به طور صحیح توسط روش های مبتنی بر فلومتریک (بررسی کمیت دقیق DNA دو رشته ایی) قابل بررسی نیست. کمیت و کیفیت DNA ورودی در آزمایش مهم است و هم چنین مقدار DNA ورودی بستگی به الزامات ورودی کیت های آماده سازی کتابخانه دارد. در تعیین توالی ژنوم (به ویژه کتابخانه هایی با قطعه درج شده بلند) به DNA با کیفیت بالا و غیر خورد شده به مقدار کافی مورد نیاز است. برای توالی یابی یک ژنوم کامل و با استفاده از کتابخانه های متفاوت، به یک میلی گرم از DNA به عنوان ماده اولیه نیاز است (6 میکروگرم برای کتابخانه های با قطعه درج شده کوتاه، 40 میکروگرم برای کتابخانه های 2-10 kb، میکروگرم برای کتابخانه های با طول بیشتر از 20 kb). کیفیت DNA باید از طریق ژل با قدرت بالا چک شود (االکتروفورز pulsed-field که قطعات با طول بیشتر از 100 kb را نشان دهد).
استانداردسازی غلظت DNA برای نمونه ها
هر نمونه DNA به مقدار استاندارد مورد نظر رقیق یا غلیظ می شود. مایع رقیق کننده باید DNA را تثبیت کرده و از آن در مقابل آسیب ها محافظت کند (مانند lowTE ). برای از بین بردن خطای پیپتینگ، استفاده از ربات دستی مایع توصیه می شود.
DNA pooling (Pool-seq)
Pooling شامل مخلوط کردن مقادیر یکنواخت از DNA مربوط به چندین فرد از یک جمعیت است. زمانی که هدف شناسایی پایه ژنتیکی یک صفت است، مجموع نمونه ها باید شامل افرادی باشند که دارای ویژگی یکسانی هستند (نه لزوماً از یک جمعیت). حداقل 50 نفر برای تهیه هر pool توصیه می شود، اما تعداد افراد بیشتر (> 100) (با فرض افزایش نسبی عمق توالی یابی) می تواند ناهماهنگی های جزئی را در نتیجه تعداد افراد کمتر به حداقل برساند و در نهایت منجر به برآورد دقیقتر فرکانس آللی شود.
هر DNA جداگانه برای رسیدن به غلظت استاندارد رقیق می شود و از طریق مرحله کمی سازی تأیید می شود. پس از نرمال سازی، مقدار مشابه/یکسان از DNAی نمونه های افراد مختلف را می توان در یک لوله واحد جمع کرد.
آماده سازی کتابخانه تعیین توالی
چندین کیت برای تهیه کتابخانه به صورت تجاری در دسترس است. نیاز به دستگاه سونیکاتور، مرحله PCR و مقدار DNA ورودی باعث تفاوت در هزینه این کیت ها می شود. تکثیر DNA با PCR زمانی مناسب است که مقادیر کمی از DNA در دسترس باشد، اما به طور کلی PCR می تواند باعث سوگیری هایی (به عنوان مثال، عدم تکثیر قطعات غنی از GC، تکثیر ترجیحی قطعات کوتاه و تکراری) در تکثیر DNA شود که منجر به پوشش ناهموار در برخی لوکوس های ژنتیکی می شود. برخی از این سوگیری ها را می توان با انجام تنظیمات پروتکل PCR (به عنوان مثال، با استفاده از تعداد کمی سیکل PCR تا حد ممکن، معمولاً 6-8) و با حذف موارد تکراری در سیلیکو (in silico) با استفاده از ابزارهای Picard یا SAMTOOLS به حداقل رساند. انواع واریانت های ساختاری کوچک (INDEL و CNV) با استفاده از خوانش های کوتاه کتابخانه های استاندارد (اندازه درج 350-550 جفت باز) تشخیص داده می شود، در حالی که تشخیص انواع واریانت های ساختاری بزرگ (شامل Mbs) ممکن است نیاز به استفاده از کتابخانه های mate-pair (~2-20Kb insert size) یا خوانش های بلند داشته باشد.
DNA استخراج شده به صورت تصادفی به توالی های کوتاه تر قطعه قطعه می شود و در دو انتهای هر قطعه، آداپتورهای خاصی متصل می شوند. سایز هر قطعه (اندازه درج) اثر مهمی در آنالیز های بعدی داده دارد. برای تعیین توالی ژنوم، معمولاً چند اندازه مختلف از قطعات درج شده برای دستیابی به اطلاعات بیشتر هنگام مونتاژ انتخاب می شود.
باید توجه شود در اغلب تکنولوژی ها از چند سیکل PCR استفاده می شود که می تواند منجر به تولید خوانش های تکراری (duplicate) شده شود. خوانش های تکرای در ارزیابی کیفیت مبتنی بر پوشش اختلال ایجاد می کنند. به همین علت خوانش های داپلیکیت شده باید قبل از مرحله مونتاژ حذف شوند. داپلیکیت ها معمولا درصد اندکی را کتابخانه هایی با قطعات درج شده با سایز کوتاه (کمتر از 500 bp) شامل می شوند اما درصد داپلیکیت ها در کتابخانه هایی با قطعات درج شده با سایز بلند (بیشتر از 10 kb) به 95 درصد میرسد.
تعیین توالی کتابخانه های DNA با توان بالا
در حال حاضر، معروف ترین فناوری برای تعیین توالی با سرعت بالا و تولید کننده خوانش های کوتاه ایلومینا است، هرچند فناوری های جدیدی در حال توسعه هستند. واکنش توالی یابی در ایلومینا در یک سیستم شیشه ای به نام فلوسل انجام می شود که هر فلوسل 8 لاین دارد. قطعه DNA به یک تک رشته تبدیل می شود و با پرایمرها در کانال توالی یابی ترکیب و یک ساختار شبیه پل برای تکثیر ایجاد می کند.dNTP بدون لیبل و آنزیم Tag پلی مراز جهت تکثیر با روش PCR پل در فاز جامد اضافه می شود. قطعه تک رشته ای به یک قطعه دو رشته ای از طریق تشکیل پل تکثیر می شود. با دناتوراسیون، تک رشته مکمل جدا و در فاصله نزدیک به سطح جامد به صورت متصل باقی می ماند. با ادامه این فرایند، میلیون ها کلاستر از آنالیت های دو رشته ایی بر روی سطح جامد فلوسل بدست می آید.
اگرچه خوانش های حاصل از دستگاه های ایلومینا دقت بیشتر از 99.5 درصد را نشان می دهد، با این حال در تمایز واریانت های ژنتیکی واقعی از آرتیفکت های تکنیکی محدودیت وجود دارد. حداقل پوشش پیشنهادی برای Pool-seq، بیشتر از 50X/pool است، هرچند برای تشخیص آلل های نادر بیشتر از 100-200X لازم است. تعداد لاین های ایلومینا در فلوسل ها به سایز ژنوم مورد بررسی، پوشش ناحیه هدف برای هر نمونه در مجموع نمونه ها و میزان محصول در فلوسل بستگی دارد. در توالی یابی ایلومینا به صورت بالقوه تنوع و اختلاف لاین به لاین وجود دارد که این مشکل با توزیع کتابخانه های بارکد دار در طول چندین لاین می تواند به حداقل برسد.
آنالیز داده ها
این مرحله بخشی از فرایند توالی یابی نیست. داده های خام به دست آمده از طریق تعیین توالی (توالی با طول چند ده باز) و ابزارهایی که این توالی های کوتاه را از طریق ابزارهای بیوانفورماتیک جمع آوری می کنند، جزء چهارچوب آنالیز کل ژنوم هستند. از طرف دیگر، این توالی ها با ژنوم رفرانس ارگانیسم مورد نظر یا توالی ژنوم گونه های مشابه همتراز می شوند و برای به دست آوردن نتایج معنی دار بیولوژیکی بیشتر تجزیه و تحلیل می شوند.
کمپانی های در زمینه توالی یابی کل ژنوم
چندین شرکت در توسعه و تجاری سازی ماشین های توالی یابی نسل بعدی (که به آنها “پلتفرم” می گویند) جهت توالی یابی کامل ژنوم متمرکز هستند. شرکت Illumina به دلیل تعداد کارایی بالای پلتفرم هایی که ارایه می دهد، محبوب ترین شرکت ارایه دهنده خدمات توالی یابی می باشد. ایلومینا دارای چندین پلتفرم است. ایلومینا HiSeq یکی از رایج ترین پلتفرم های تعیین توالی در آزمایشگاها هست، از جمله موسسات تحقیقاتی بزرگ و شرکت هایی که خدمات تعیین توالی را برای کلینیک ها و آزمایشگاه های تحقیقاتی و آزمایشگاه های آسیب شناسی ارائه می دهند.
ایلومینا
در این روش از نوکلئوتیدهای اصلاح شده ای استفاده می شود که به محض اضافه شدن به انتهای رشته پلی نوکلئوتید در حال ساخت، باعث توقف ادامه سنتز زنجیره جدید شود. هریک از انواع نوکلئوتیدهای خاتمه دهنده با یک مولکول فلوئوفور نشاندار شده اند به طوریکه می توان هر یک از چهار نوکلئوتید اضافه شده در هر مرحله را نسبت به یکدیگر افتراق داد. پس از شناسایی نوکلئوتید اضافه شده، عامل شیمیایی بلاکه کننده همراه با فلوئوفور از انتهای ’3 نوکلئوتید برداشته می شود و نوکلئوتید استاندارد بجا می ماند؛ بنابراین ساخت رشته DNA از سرگرفته می شود و به همین ترتیب نوکلئوتید بعدی اضافه و شناسایی می شود. توالی یابی به روش خاتمه برگشت پذیر با کتابخانه های تثبیت شده بر روی اسلاید، توانایی خوانش توالی های نسبتا کوتاه، با حداکثر 300bp را دارد اما با انجام تعداد زیادی واکنش موازی 2000bp توالی را در هر بار اجرا می توان بدست آورد. این تکنولوژی تحت عنوان توالی یابی ایلومینا (نام شرکت ارائه دهنده) نیز شناخته می شود.
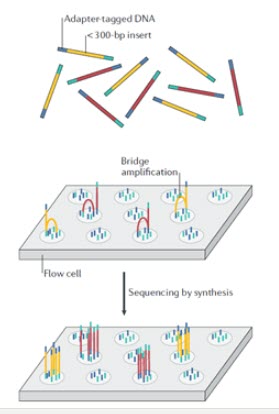
در تکنولوژی ایلومینا از روش “پل PCR ” استفاده می شود که کلاسترهای کلون شده از قطعات تکثیر شده متصل به سطح شیشه ایی تولید می شود. در این روش، قطعات DNA (زرد و قرمز) به آداپتورها (آبی و سبز) پیوند داده می شوند (شکل 7). آداپتورها شامل شناسه های مولکولی منحصر به فرد و توالی های مکمل الیگونوکلئوتیدی هستند که به سطح فلوسل ها متصل می شوند. قطعات برچسب زده شده با آداپتور بر روی فلوسل بارگذاری می شوند و این قطعه DNA-آداپتور از طریق توالی آداپتورها به الیگونوکلئوتیدهایی که سطح فلوسل را می پوشانند، هیبرید می شوند. پس از اتصال قطعات DNA، تولید کلاستر آغاز می شود، جایی که هزاران نسخه از هر قطعه طی فرآیندی به نام تکثیر پل تولید می شود. در این فرایند، بعد از سنتز یک رشته، آداپتور انتهای مولکول به یک الیگونوکلئوتید دیگر در فلوسل هیبرید می شود. آنزیم پلیمراز نوکلئوتیدها را برای ساختن پل های دو رشته ای از مولکول های DNA پشت سرهم قرار می دهد و سپس به دنبال دناتوراسیون، قطعات DNA تک رشته ای به سطح فلوسل متصل باقی می مانند. این فرایند بارها و بارها تکرار می شود و چندین میلیون خوشه متراکم از DNA دو رشته ای ایجاد می کند. پس از تقویت پل، رشته های DNA معکوس جدا و شسته می شوند و تنها رشته های فوروارد باقی می مانند. سپس، تعیین توالی توسط سنتز آغاز می شود، که در آن تری فسفات نوکلئوزید با نشان فلورسنت در رشته چرخه DNA تازه سنتز شده در هر چرخه گنجانده می شود. پس از اتصال، لیزر فلوروفور روی رشته را تحریک می کند، که بدنبالش یک سیگنال فلورسانس مشخص و مربوط به بازمتصل شده را منتشر می کند.
سیستم های Pacific Bioscience
یکی از روش های نسل سوم تعیین توالی، توالی یابی تک مولکول DNA در زمان واقعی (SMRT) است. نام دیگر این سیستم PacBio RSII است که در سال 2010 توسعه یافت و به عنوان نخستین روش توالی یابی DNA برای توالی یابی مولکول های تکثیر نشده منفرد در زمان واقعی شناخته شد. در این تکنولوژی یک سیستم نوری پیچیده تحت عنوان ZMW برای مشاهده کپی برداری از یک مولکول DNAی الگو استفاده می شود و اضافه شدن هر نوکلئوتید به وسیله برچسب فلورسنت متصل شده به آن شناسایی می شود (شکل 8).
توالی یابی تک مولکول در زمان واقعی، در یک ساختار حاوی 150000 چاهک ریز رخ می دهد که موج بر مد صفر نامیده می شود. یک مولکول DNA پلی مراز منفرد به انتهای هر چاهک متصل می شود. ZMW یک سیستم نوری بسیار دقیق است و به اندازه کافی کوچک است تا تنها یک نوکلئوتید را که توسط DNA پلیمراز به رشته در حال ساخت اضافه می شود مشاهده کند. در این سیستم نیازی به بلاک کردن کربن `3 نوکلئوتید نیست، بلکه سیگنال فلورسنت شناسایی می شود و برچسب بلافاصله پس از اضافه شدن نوکلئوتید به زنجیره در حال ساخت برداشته می شود، بنابراین سنتز زنجیره بدون توقف ادامه می یابد. با این روش توالی یابی، توالی هایی با حداکثر 20000 جفت باز توالی یابی شده اند. بنابراین، به طور خلاصه، هر یک از چهار باز به یکی از چهار رنگ مختلف فلورسنت متصل شده است. هنگامی که یک نوکلئوتید توسط DNA پلیمراز به رشته در حال ساخت اضافه می شود، برچسب فلورسنت جدا می شود و از منطقه مشاهده ZMW خارج می شود، جایی که فلورسانس آن دیگر قابل مشاهده نیست. یک آشکارساز سیگنال فلورسنت نوکلئوتید را تشخیص می دهد.
PacBio RSII، انجام خوانش های بسیار بلند را محقق می سازد و زمان آماده سازی نمونه و مدت زمان انجام فرایند کوتاه بوده که باعث می شود کل فرایند در یک روز انجام شود. خطاها تصادفی هستند. هزینه کار برای هر باز بسیار بیشتر از سایر سیستم های رقابتی است. با این حال خوانش های بلند، این روش را برای توالی یابی de novo ژنوم های باکتریایی یا ویروسی کوچک ایده آل می سازد.
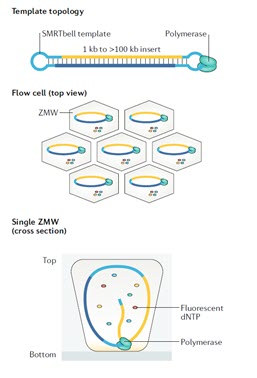
شکل 8. توالی یابی مولکول منفرد در زمان واقعی با استفاده از سیستم SMRT. (A) الگو برای این توالی یابی، DNA دو رشته ایی همراه با سنجاق سرهای (hairpin) تک رشته ای متصل به دو انتها است. یک پرایمر توالی یابی (قرمز) به یکی از ساختارهای سنجاق سری متصل می شود. آنزیم پلی مراز جابجا شونده روی رشته، به طور پیوسته روی الگو حرکت می کند و نسخه های کنکاتامری از کل توالی را ایجاد می کند. (B) پلی مراز به ته چاهک متصل شده است. چهار dNTP نشاندار شده با فسفات نیز به طور آزادانه پراکنده شده اند. (C) وقتی یک نوکلئوتید وارد رشته در حال سنتز می شود، نشانه فلورسنت آن در ته چاهک بسیار بیشتر از dNTPهای آزاد باقی می ماند.
فناوری نانوپور
در سال های اخیر، فناوری نانوپور (سیستم منافذ نانو آکسفورد) در زمینه علوم زیستی و تحقیقات زیست پزشکی از اهمیت فزاینده ای برخوردار شده است. با قرار دادن یک حفره در مقیاس نانو در یک غشای نازک و سپس اندازه گیری سیگنال الکتروشیمیایی، می توان از فناوری نانو منفذ برای بررسی اسیدهای نوکلئیک و سایر مولکول های زیستی استفاده کرد. یکی از موفق ترین کاربردهای فناوری نانوپور، فناوری نانوپورآکسفورد است که آغاز نسل چهارم فناوری تعیین توالی ژن است.
همه دستگاه های توالی یابی آکسفوردنانوپور از فلوسل هایی استفاده می کنند که شامل مجموعه ای از حفره های کوچک (نانو حفره ها) است که در یک غشاء مقاوم در برابر الکتریکی قرار گرفته اند. هر نانو حفره الکترود مخصوص خود را دارد که به یک کانال و تراشه حسگر متصل شده است، که جریان الکتریکی را که در نانو حفره عبور می دهد اندازه گیری می کند. هنگامی که یک مولکول DNA از یک نانو حفره عبور می کند، جریان مختل می شود تا “حالت موج دار” مشخص ایجاد شود. سپس با استفاده از الگوریتم های فراخوانی بازها برای تعیین توالی DNA یا RNA در زمان واقعی رمزگشایی می شود.
به طور خلاصه، دو استراتژی اصلی تعیین توالی ژنوم بر اساس خوانش های کوتاه و بلند وجود دارد: تعیین توالی ژنوم خوانش های کوتاه با استفاده از تکنولوژی ایلومینا که خوانش های کوتاه با انتهای جفت شده (150جفت باز) تولید می کنند و میزان خطای این تکنولوژی در محدوده 0.5-0.1 درصد است. نوع دوم، تعیین توالی ژنوم با خوانش های بلند با استفاده از تکنولوژی های Pacific و Oxford Nanopore انجام می شود و خوانش هایی با طول 100-10 کیلوبایت و میزان خطای 15-10 درصد را فراهم می کنند. عواملی مثل هزینه، آسانی و صحت کار باعث شده که در بیشتر مطالعات ژنتیک انسانی از تعیین توالی ژنوم با خوانش های کوتاه (با استفاده از پلتفرم های ایلومینا Hiseq یا Novaseq) استفاده شود.
آنالیز داده های توالی یابی ژنوم
کنترل کیفیت توالی های خام توالی یابی ژنوم
داده های خام تعیین توالی در فرمت fastq فراهم میشوند. کنترل کیفیت این داده ها با ابزار FASTQC انجام می شود. بعد از کنترل کیفی، بازهایی با کیفت پایین (PHRED quality score <20) پیرایش و توالی های آداپتور با استفاده از ابزارهایی مانند TRIMMOMATIC یا CUTADAPT حذف می شوند.
الاین کردن با ژنوم رفرانس
خوانش های با کیفیت بالا به ژنوم رفرانس الاین می شوند. الگوریتم های مختلفی برای خوانش هایی با طول کوتاه استفاده می شود از جمله BWA و Bowtie2. فرمت فایل ایجاد شده به صورت SAM و دارای چندین گیگابایت حجم است. با استفاده از ابزار Picard فایل SAM به فایل فشرده و اجرایی BAM تبدیل می شود. مرحله بعد مرتب کردن خوانش ها، حذف داپلیکیت ها، اضافه کردن Read Groups و ایندکسینگ فایل BAM است تا فایل نهایی آماده ی variant calling شود.
کنترل کیفی خوانش های الاین شده
فایل BAM با نرم افزار IGV قابل مشاهده است که نشان دهنده نواحی با پوشش پایین و بالا، سوگیری رشته، ناهماهنگی در الایمنت اطراف ایندل ها و نواحی تکراری و موارد دیگر می شود. در صورتی که خطاهای همترازی یا الاینمنت داشته باشیم باید تصحیح صورت بگیرد و خوانش های الاین شده صحیح و با کیفیت انتخاب شوند تا از تعداد واریانت های کاذب در مرحله variant calling کاسته شود. معیارهای آماری در بررسی کنترل کیفی مرحلع الایمنت توسط ابزارهای SAMTOOLS و QUALIMAP به دست می آید. این معیارها شامل موارد زیر هستند: میانگین عمق پوشش، توزیع سایز خوانش ها، تعداد خوانش های الاین شده، تعداد خوانش ها به صورت جفت، تعداد خوانش های الاین شده مبهم و منفرد.
الاینمنت مجدد Indels
الاینمنت مجدد Indels (بستگی به نوع SNP caller دارد). الایمنت دقیق آرتیفکت ها در اطراف ایندل ها توسط پارامترهای الایمنت گلوبال برطرف نمی شود. زمانی که الگوریتم های site-based SNP calling مانند SAMtools یا GATK-UNIFIEDENOTYPER استفاده می شود لازم است الایمنت مکانی ایندل ها انجام شود. این مرحله در مورد Haplotype-based callers مانند FREEBAYES و GATK-HAPLOTYPEVALLER لازم نمی باشد.
کالیبراسیون پایه
اسکورهای کیفیت بدست آمده از دستگاه های تعیین توالی برای هر باز اغلب با خطا همراه هست. الگوریتم های SNP calling و تعیین ژنوتایپ نیز اسکورهای کیفیت ارائه می دهند، از جمله پکیج کالیبراسیون اسکور کیفیت باز (BQSR) که در GATK وجود دارد.
تشخیص واریانت های مختلف
ابزارهای خاصی برای بررسی و تشخیص انواع مختلف واریانت های ژنتیکی استفاده می شود. از جمله GATK-HAPLOTYPECALLER، SAMTOOLS یا FREEBAYES که برای SNP calling استفاده می شود. فرمت فایل ایجاد شده vcf است که حاوی پلی مورفیسم ها می باشد.
کنترل کیفی واریانت ها
SNP ها با کیفیت پایین باید حذف شوند که برای اینکار از الگوریتم VQSR با فیلتر سخت استفاده می شود. فایل vcf بعد از کنترل کیفی برای مراحل بعد استفاده می شود.
مرحله annotation واریانت ها
بزنامه های VCFANNO، ANNOVAR، SNPEFF برای اَنوتیت کردن واریانت ها در فایل vcf استفاده می شوند.
تایید واریانت ها
روش های مبتنی بر PCR و تعیین توالی سنگر در تایید SNP ها و واریانت های ساختاری استفاده می شوند.
پایگاه های داده و ذخیره سازی داده های توالی یابی کل ژنوم
پایگاه داده SRA
دیتابیس SRA منبع عمومی برای داده های توالی یابی DNA، مخصوصا خوانش های تولید شده با روش های تعیین توالی با قدرت بالا می باشد. این آرشیو قسمتی از دیتابیس های INSDC، NCBI، EBI و DDBJ است. این دیتابیس ها خدمات ارزشمندی را برای بایگانی کردن طیف وسیعی از داده های تعیین توالی فراهم کرده اند. این آرشیو توسط NCBI در سال 2007 منتشر شد و منبعی از داده های تولید شده توسط RNA-seq می باشد و هم چنین منبعی از مطالعات بزرگی چون پروژه میکروبیوم انسانی و پروژه 1000 ژنوم می باشد.
پایگاه داده GSA
دیتابیس GSA یک منبع ذخیره برای تهیه آرشیو از داده های خام خوانش های تعیین توالی در مرکز داده ژنومیک ملی چین است. دسترسی برای عموم جامعه علمی جهان فراهم است و از طرفی از سراسر جهان داده دریافت می کنند و مسئولیت نگهداری و کنترل کیفی داده های دریافت شده را بر عهده دارند. تا سال 2015، 359017 آزمایش و 8756 ترابایت فایل دریافت شده است.
پایگاه داده GDC
دیتابیس GDC اطلاعات جامعی را در مورد سرطان فراهم می کند. GDC چندین دیتابیس مرتبط با ژنوم سرطان مانند CCG ، TCGA و TARGET را در بر می گیرد.
GDC با شناسایی تغییرات در سلول های سرطانی که نقش مهمی در پیشرفت سرطان دارند به محققین در زمینه ژنومیک سرطان کمک می کند. از طریق پایگاه GDC، محققان می توانند از داده های موجود در GDC برای شناسایی عوامل ایجاد کننده در سرطان با فرکانس بالا و پایین استفاده کنند مانند:
موتاسیون ها: دسترسی به داده های DNA توالی یابی شده امکان شناسایی موتاسیون ها را فراهم می کند. در این مرحلهخروجی هایی با فرمت vcf و maf ایجاد می شود که جهش های سوماتیک مانند جهش های نقطه ای، جهش های بی معنی و درج و حذف ها در آن گزارش می شود.
تغییرات تعداد کپی: این دیتابیس دسترسی به شناسایی CNV ها را فراهم میکند.
بررسی کمیت بیان ژن: GDC دسترسی به داده های بیان ژن از جمله mRNA و miRNA را فراهم می کند. فرمت این فایل ها TSV است.
مدیفیکاسیون های بعد از ترجمه (post translational modification (PTM)): دسترسی به داده های توالی mRNA باعث شناسایی تغییرات اسپلایسینگ بعد از ترجمه می شود.
اطلس ژنوم سرطان و کنسرسیوم بین المللی ژنوم سرطان
اطلس ژنوم سرطان (TCGA) و کنسرسیوم بین المللی ژنوم سرطان (ICGC) به ترتیب دارای اطلاعات توالی اگزوم و توالی ژنوم می باشند. کنسرسیوم بین المللی ژنوم سرطان (ICGC) برای هماهنگی مطالعات ژنوم سرطان در مقیاس بزرگ در تومورهایی از 50 نوع مختلف سرطان و/یا زیرگونه هایی از سرطان که اهمیت بالینی و اجتماعی در سراسر جهان دارند، راه اندازی شد. بیش از 25000 داده مرتبط با سرطان در سطوح ژنومی، اپی ژنومی و ترانسکریپتومی، مجموعه جهش های سرطان زا و ردپایی از تأثیرات جهش زا را آشکار می کنند و هم چنین مطالعات بالینی مرتبط با پیش آگهی، مدیریت درمان و توسعه درمان های جدید سرطان را ممکن می سازند. اطلس ژنوم سرطان (TCGA)، برنامه ای مهم در ژنومیک سرطان است که بیش از 20000 سرطان اولیه را از نظر مولکولی بررسی کرده است. این تلاش مشترک بین NCI و موسسه ملی ژنوم انسانی در سال 2006 آغاز شد و محققان رشته های مختلف و موسسات متعدد را گرد هم آورد. در طول دوازده سال آینده، TCGA بیش از 2.5 پتابایت اطلاعات ژنومی، اپی ژنومی، ترانسکریپتومی و پروتئومی تولید خواهد کرد. این داده ها، که قبلاً منجر به بهبود توانایی ما در تشخیص، درمان و پیشگیری از سرطان شده است، برای استفاده همه افراد جامعه تحقیقاتی در دسترس عموم قرار می گیرد.
پایگاه داده GenomAD
دیتابیس GenomAD منبع اطلاعاتی داده های تعیین توالی اگزوم و ژنوم با استفاده از پروژه های تعیین توالی در سطح وسیع است. داده های این دیتابیس شامل موارد زیر است:
مجموعه دیتا v2 شامل 125748 توالی اگزوم و 15708 توالی ژنومی افراد غیرخویشاوند که به ژنوم رفرانس GRCh37/hg19 الاین شده اند.
مجموعه دیتا v3 شامل 76156 توالی ژنوم که با ژنوم رفرانس GRCh38 همتراز شدند.
پروژه 1000 Genomes
پروژه 1000 ژنوم با استفاده از نمونه های افرادی که سالم معرفی شده اند فهرستی از تغییرات ژنتیکی مشترک انسان ها را ایجاد کرده است. در اوایل سال جاری(2021)، مرکز ژنوم نیویورک (NYGC) داده های با پوشش بالا (30X) را برای 698 نمونه دیگر، علاوه بر مجموعه نمونه های پروژه 1000 ژنوم منتشر کرد. این 698 نمونه مربوط به مجموعه اصلی 2،504 نمونه ایی است که قبلاً توسط NYGC تعیین توالی شده بود. 2،504 نمونه، مجموعه ای از نمونه های غیرخویشاوند و غیرمرتبط با یکدیگر هستند. در کل، تعداد کل نمونه های با پوشش بالا توسط NYGC به 3202 مورد می رسد.
پروژه UK10K
پروژه UK10K، محققان در بریتانیا و خارج از آن را قادر می سازد تا ارتباط بین تغییرات ژنتیکی با فرکانس آللی نادر و بیماری های انسانی ناشی از تغییرات مضر پروتئین های بدن را بهتر درک کنند. اگرچه صدها ژن دخیل در ایجاد بیماری قبلاً شناسایی شده است، اما اعتقاد بر این است که ژن های بیشتری هستند که کشف نشده اند. پروژه UK10K با کشف کد ژنتیکی 10 هزار نفر با جزئیات بسیار دقیق تر از قبل به کشف آنها کمک می کند. نمونه هایی که در این مطالعه استفاده شدند شامل 4000 نمونه توالی یابی کل ژنوم، 3000 نمونه توالی یابی شده اگزوم مربوط به نمونه های سیستم عصبی، 2000 نمونه تعیین توالی اگزوم مربوط به چاقی و 1000 نمونه تعیین توالی اگزوم مربط به بیماری های نادر می باشد.
پروژه Genome 100kb
این پروژه برای توالی 100000 داده ژنومیک طراحی شده است و از این تعداد حدود 85000 بیمار مراجعه کننده به سیستم ملی سلامت (NHS142) همراه با یک بیماری نادر یا سرطان شرکت کرده اند. این پروژه خدمات پزشکی ژنومی جدیدی را برای بیماران مراجعه کننده به NHS ایجاد می کند از جمله نحوه مراقبت از مردم را تغییر می دهد و تشخیص پیشرفته و درمان های شخصی را برای همه کسانی که به آنها نیاز دارند فراهم می کند.
کاربردها
مزیت اصلی تعیین توالی ژنوم، بررسی کل ژنوم (نواحی کد کننده و غیر کد کننده و میتوکندریایی) به صورت یک مرحله ایی است و حجم زیادی از داده ها را در مدت زمان کوتاهی برای مونتاژ ژنوم های جدید ارائه می دهد. بدین معنی که در یک مرحله، هر واریانتی از نوع تک نوکلئوتید تا یک تغییر بزرگ در سطح تعداد کپی یا جابجایی و تغییرات ساختاری تشخیص داده می شود.هم چنین جهش های موجود در توالی های تنظیمی که در فضای خارج ژنی یا درون نواحی مرکزی اینترون ها قرار گرفته اند را تشخیص می دهند.
این اطلاعات به دست آمده از توالی یابی ژنوم ارزشمند است و به تشخیص بیماری مونوژنیک (مانند تالاسمی) یا بررسی استعداد به بیماری های پلی ژنیک مانند دیابت تایپ دو کمک می کند. علاوه بر تشخیص جهش ها در افراد مبتلا به تشخیص ناقلین بیماری های ژنتیکی و سپس مشاوره ی ژنتیک صحیح نیز کمک می کند. همچنین در تحقیقات سرطان جهت شناسایی جهش های توموری و بررسی درمان های جدید بیولوژیکال مفید است.
محدودیت ها
مشکل اصلی مربوط به توالی یابی کل ژنوم حجم زیادی از اطلاعات تولید شده است، که باید تجزیه و تحلیل و ارزیابی شود تا مشخص شود چه داده ایی مهم است یا چه داده ایی مهم نیست. اگرچه دانش ما در زمینه ژنومیک در حال افزایش است، با این وجود نقش بسیاری از ژن ها هنوز نامشخص است و هم چنین تعداد زیادی از واریانت ها در سرتا سر ژنوم هنوز به عنوان خوش خیم یا بیماری زا مشخص نشده اند. این بدان معناست که اگرچه توالی یابی کل ژنوم می تواند حجم زیادی از داده ها را تولید کند، اما اکثر آنها ممکن است گمراه کننده یا بی فایده باشند و به آنالیز های بیوانفورماتیکی بسیار وسیع تری نیاز باشد.
حجم زیادی از داده های تولید شده توسط توالی یابی کل ژنوم نه تنها نیاز به تجزیه و تحلیل دارد، بلکه باید ذخیره شود. این به خودی خود برخی از چالش ها را ایجاد می کند، به عنوان مثال، نه تنها به ظرفیت و هزینه زیاد برای این کار، بلکه با حریم خصوصی داده ها، که می تواند مسائل اخلاقی را برای شرکت های بیمه و اعضای خانواده ایجاد کند نیز مواجه هستیم.
توالی یابی کل ژنوم ممکن است یافته های ثانویه ناخواسته ایی را کشف کند. بنابراین، ممکن است تشخیص بیماری ژنتیکی غیر قابل درمان و با شروع دیررس در سال های آینده را نشان دهد (مانند بیماری هانتینگتون). این موضوع می تواند تأثیر روانی منفی بر فرد و همچنین اعضای خانواده داشته باشد و علاوه بر این می تواند ارتباطات خانوادگی را تحت تاثیر قرار دهد، همانطور که سایر اعضای خانواده ممکن است مایل به آگاهی از چنین اطلاعاتی نباشند، دیگران ترجیح می دهند بدانند. علاوه بر این، این سوال را مطرح می کند که در صورت وجود چنین داده و اطلاعاتی، کدام نتایج باید به خانواده ها اعلام شود.